

Фото: Kmpzzz/FOTODOM/Shutterstoсk
Журнал "Медицинский совет" №15/2025
DOI: 10.21518/ms2025-399
С.И. Мозговой1, О.В. Гаус1, М.А. Ливзан1, Д.С. Бордин2,3,4, В.А. Рубцов1, И.А. Рыльская1
1 Омский государственный медицинский университет; 644099, Россия, Омск, ул. Ленина, д. 12
2 Московский клинический научный центр имени А.С. Логинова; 111123, Россия, Москва, ул. Новогиреевская, д. 1, корп. 1г
3 Российский университет медицины (РосУниМед); 127006, Россия, Москва, ул. Долгоруковская, д. 4г
4 Тверской государственный медицинский университет; 170100, Россия, Тверь, ул. Советская, д. 4
Инфекция Helicobacter pylori (H. pylori) и аутоиммунное воспаление слизистой оболочки желудка признаны ведущими этиологическими факторами хронического атрофического гастрита. При этом механизмы формирования и прогрессирования атрофии слизистой оболочки с риском развития рака желудка при данных типах гастритов очень гетерогенны, требуют более глубокого изучения данного вопроса для разработки последующего персонифицированного подхода к ведению больных. Так, в последние годы появились данные, свидетельствующие о том, что аутоиммунный гастрит (АИГ) без сопутствующей инфекции H. pylori и без признаков значительного воспаления другой этиологии характеризуется относительно доброкачественным течением и низкой вероятностью неопластической трансформации слизистой оболочки желудка в рак желудка. Однако появление данных о возможности инфекции H. pylori индуцировать аутоиммунный процесс в отношении собственных париетальных клеток с развитием атрофии слизистой оболочки тела желудка делает актуальным проблему ведения и определения рисков канцерогенеза у больных хроническим гастритом смешанной этиологии. Целью данного научного обзора является систематизация имеющихся данных в отношении АИГ, механизмов развития атрофии и ассоциированных с ней рисков канцерогенеза как при изолированном аутоиммунном воспалении, так и в условиях персистирующей или предшествующей инфекции H. pylori («чистый» АИГ), персистирующей инфекции H. pylori или в период после эрадикационной терапии. Особое внимание уделено феномену молекулярной мимикрии между антигенами H. pylori и эпитопами собственных париетальных клеток желудка при развитии гастрита смешанной этиологии.
Для цитирования: Мозговой СИ, Гаус ОВ, Ливзан МА, Бордин ДС, Рубцов ВА, Рыльская ИА. Аутоиммунный гастрит и инфекция Helicobacter pylori: молекулярные механизмы взаимосвязи. Медицинский Совет. 2025;(15):16-26. https://doi.org/10.21518/ms2025-399
Конфликт интересов: автор заявляет об отсутствии конфликта интересов.
Autoimmune gastritis and Helicobacter pylori infection: Molecular mechanisms of relationship
Sergei I. Mozgovoi1, Olga V. Gaus1, Maria A. Livzan1, Dmitry S. Bordin2,3,4, Vyacheslav A. Rubtsov1, Irina A. Rylskaia1
1 Omsk State Medical University; 12, Lenin St., Omsk, 644099, Russia
2 Loginov Moscow Clinical Scientific Center; 1, Bldg. 1, Novogireevskaya St., Moscow, 111123, Russia
3 Russian University of Medicine (ROSUNIMED); 4g, Dolgorukovskaya St., Moscow, 127006, Russia
4 Tver State Medical University; 4, Sovetskaya St., Tver, 170100, Russia
For citation: Mozgovoi SI, Gaus OV, Livzan MA, Bordin DS, Rubtsov VA, Rylskaia IA. Autoimmune gastritis and Helicobacter pylori infection: Molecular mechanisms of relationship. Meditsinskiy sovet = Medical Council. 2025;(15):16-26. (In Russ.) https://doi.org/10.21518/ms2025-399
Conflict of interest: the author declares no conflict of interest.
Введение
Инфекция Helicobacter pylori (H. pylori) и
аутоиммунное воспаление слизистой оболочки желудка относятся к ведущим
этиологическим факторам развития атрофического гастрита [1]. Следует отметить,
что механизмы формирования воспаления, как и проканцерогенный потенциал,
различаются, что делает крайне актуальным сопоставление факторов патогенеза как
в условиях изолированного действия каждого из этиологических факторов, так и
при сочетанном их воздействии.
Целью данного научного обзора является
систематизация имеющихся данных в отношении аутоиммунного гастрита (АИГ), механизмов
развития атрофии и ассоциированных с ней рисков канцерогенеза как при
изолированном аутоиммунном воспалении, так и в условиях персистирующей или
предшествующей инфекции H. pylori. Отдельное
внимание уделено феномену молекулярной мимикрии между антигенами
H. pylori и эпитопами
собственных париетальных клеток желудка при развитии гастрита смешанной
этиологии.
Был проведен систематический поиск статей в базах данных PubMed/MEDLINE, Embase и Google
Scholar. Для поиска статей использовались комбинации из следующих ключевых слов
на основе предметной медицинской рубрики (MeSH), включая «аутоиммунный гастрит»,
«H. pylori-ассоциированный
гастрит», «гастрит после эрадикации H. pylori», «молекулярная мимикрия H. pylori». Критерии отбора статей для данного обзора были
сформированы в соответствии с международными рекомендациями, изложенными в
стандарте PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) [2]. В анализ включались статьи, соответствующие
тематике обзора, опубликованные на английском языке в авторитетных и
высокорейтинговых изданиях до мая 2025 г., в т. ч. отдельные
клинические наблюдения, оригинальные исследования, систематические обзоры и
метаанализы с ясным и корректным описанием методологического подхода,
обеспечивающего воспроизводимость и валидность полученных данных. После
применения критериев отбора в данный обзор было включено в общей сложности 85
статей.
Аутоиммунный
гастрит – хроническое аутоиммунное заболевание с поражением тела и дна желудка,
характеризующееся иммунным ответом, направленным на париетальные клетки и
внутренний фактор. Распространенность АИГ в популяции, по данным ряда авторов,
составляет от 1 до 8%, чаще встречается у женщин в сравнении с мужчинами в
соотношении 3:1 [1].
Аутоиммунное
воспаление слизистой оболочки желудка формируется при взаимодействии генетических
и средовых факторов.
Генетические
факторы риска АИГ изучены недостаточно. Первые исследования были сосредоточены
на поиске ассоциаций между риском АИГ и гаплотипами системы человеческого
лейкоцитарного антигена (HLA), основным эффектом которых является презентация
антигенов иммунным клеткам [3]. Так, B. Ungar et al. описали повышенную частоту гаплотипов HLA-B8, HLA-B18 и HLA-Bw*15 у пациентов с АИГ и пернициозной анемией [4]. Другое исследование выявило повышенную
частоту HLA-DR2 или HLA-DR4 и HLA-DR4 или HLA-DR5 у пациентов с пернициозной анемией [5]. Гетерогенность гаплотипов HLA в разных
клинических подгруппах может отражать генетическую гетерогенность пернициозной
анемии и, возможно, АИГ. В литературе имеется 2 исследования в отношении
предрасположенности к АИГ в зависимости от вариаций генов HLA [6, 7]. В итальянской популяции среди 89
пациентов с АИГ распространенность HLA-DRB1*03 и HLA-DRB1*04 была выше по сравнению со здоровыми лицами (28,1%
против 15,9%, p = 0,01,
и 25,8% против 14,4%, p = 0,01 соответственно) [6]. Кроме того, HLA-DRB1*03 и HLA-DRB1*04 были связаны с наличием кишечной метаплазии (р < 0,01).
В финской популяции среди 12 пациентов с АИГ и тяжелой атрофией слизистой
оболочки тела желудка значительно чаще выявлялись HLA-DRB1*04 и DQB1*03 (83%), чем в общей популяции (58% против 28%, р = 0,045,
и 83% против 51%, р = 0,034 соответственно) [7].
Указанные гаплотипы HLA также
часто связаны с другими аутоиммунными состояниями, тем самым поддерживая общий HLA-зависимый путь развития аутоиммунной
патологии [3].
В
число изучаемых генов, предположительно ассоциированных с АИГ, входят гены ATP4A и ATP4B, участвующие в синтезе субъединиц
α и β специфического трансмембранного фермента
водородно-калиевой аденозинтрифосфатазы (H+/K+-АТФазы) и, следовательно, в поддержании секреции соляной кислоты [8]; ген
AIRE, кодирующий фактор транскрипции в тимусе,
который играет ключевую роль в уничтожении аутореактивных Т-клеток [9]; ген BACH2, кодирующий один из двух факторов
транскрипции семейства Bach, которые вовлечены в контроль иммунного ответа
посредством транскрипционной репрессии целевых генов [10]; гены SLC4A2, SLC26A7 и SLC26A9, кодирующие синтез соответствующих членов А2, А7 и А9 семейства
растворенных носителей 4 и 26, играющих ключевую роль в модуляции
кислотно-щелочного баланса и обмена H+/K+ в париетальных клетках [11]. Метаанализ полногеномного
исследования 2 166 пациентов с пернициозной анемией и 659 516 лиц контрольной
группы из популяционных биобанков выявил следующие гены, связанные с
пернициозной анемией: протеин-тирозинфосфатазу типа 22 (PTPN22), полирибонуклеотид-нуклеотидилтрансферазу-1
(PNPT1), гаплотип главного комплекса
гистосовместимости класса II (MHC II) DQ бета 1 (HLA-DQB1), субъединицу альфа-рецептора интерлейкина-2 (IL2RA) и AIRE [12].
Среди
других генетических факторов, играющих важную роль в развитии и прогрессировании
воспалительных изменений слизистой оболочки желудка, активно изучаются гены,
связанные с иммунным ответом хозяина, в частности гены, кодирующие экспрессию Toll-подобных рецепторов (TLR) [13]. TLR являются важной частью врожденной
иммунной системы и играют ключевую роль в распознавании различных экзогенных
патоген-ассоциированных молекулярных паттернов (PAMP) бактерий и эндогенных
молекулярных паттернов, связанных с повреждением клеток-хозяина (DAMP). У людей
идентифицировано десять типов TLR, некоторые из них присутствуют в
эпителиальных клетках желудка как в нормальных, так и в патологических
условиях. Как правило, считается, что TLR вызывают и усиливают воспаление и
клеточную пролиферацию при инфекциях и в процессе онкогенеза [14], однако TLR9 может проявлять как
провоспалительные, так и противовоспалительные функции в зависимости от
конкретного микроокружения [15]. Основная
биологическая роль TLR заключается в запуске
нисходящих внутриклеточных сигнальных путей, таких как путь ядерного фактора
каппа B (NF-kB) и инфламмасомы, что
приводит к активации генов, ответственных за выработку провоспалительных
цитокинов, хемокинов, ко-стимулирующих и антиген-презентирующих молекул,
которые инициируют специфические иммунные ответы [16].
Установлено, что TLR играют важную роль в иммунном ответе на инфекцию H. pylori и могут влиять на процессы канцерогенеза [17].
Различные компоненты H. pylori, такие как липополисахарид (ЛПС),
пептидогликан и бактериальные нуклеиновые кислоты, служат в качестве PAMP,
распознаваемых определенными TLR. Например, TLR2 и TLR4 являются ключевыми рецепторами, которые идентифицируют
пептидогликан и ЛПС клеточной стенки H. pylori [18]. Взаимодействие
ЛПС H. pylori с TLR2 и TLR4 ассоциировано с высокой
продукцией провоспалительных цитокинов: интерлейкина-8 (ИЛ-8), интерлейкина-1β
(ИЛ-1β) и фактора некроза опухоли альфа
(ФНО-α), которые играют центральную роль в инициировании и поддержании
воспалительного ответа [19]. Кроме того, воспаление, опосредованное TLR, может модулировать активацию и
дифференциацию подгрупп Т-хелперных клеток (Th), таких как Th1 и Th17, участвующих в иммунной защите от H. pylori [20]. Таким образом, вполне вероятно, что генетика TLR определяет
выраженность иммунных и воспалительных реакций в слизистой оболочке желудка организма-хозяина
в ответ на инфицирование H. pylori.
Помимо
генетической предрасположенности, к немодифицируемым
факторам риска, предрасполагающим к развитию АИГ, относится женский пол. Данный
факт связан не только с Х-хромосомой, но и с широким спектром воздействия
половых гормонов на иммунную систему и органы-мишени. Половые гормоны
регулируют молекулярные механизмы во врожденной и адаптивной иммунных системах.
Сложные взаимодействия гормонов и факторов окружающей среды у генетически
восприимчивых лиц приводят к нарушению регуляции иммунного ответа, что ведет к
иммуноопосредованным заболеваниям, включая АИГ [21].
К средовым факторам риска
АИГ относятся особенности питания, перенесенные вирусные и бактериальные
инфекции. Предполагается связь
между высококалорийной диетой, богатой простыми углеводами, рафинированными продуктами,
насыщенными жирами и красным мясом на фоне низкого содержания полиненасыщенных омега-3
жирных кислот и растительной клетчатки, с повышенным риском аутоиммунных
заболеваний [22]. Наряду с особенностями питания, рассматривается также роль
инфекционных агентов, среди которых наиболее изучена взаимосвязь с
аутоиммунной патологией таких вирусов, как цитомегаловирус (ЦМВ), вирус
простого герпеса, вирус Эпштейна – Барр. Так, на сегодняшний день установлена
связь между латентной ЦМВ-инфекцией и развитием аутоиммунного тиреоидита,
выявлена высокая корреляция между носительством герпес-вируса, ЦМВ и развитием
аутоиммунного антифосфолипидного синдрома у женщин с невынашиванием
беременности [23]. Немаловажную роль в потенцировании развития АИГ отводится и инфекции
H. pylori. В рамках существующей
гипотезы о молекулярной мимикрии H. pylori
способен выступать триггером в запуске аутоиммунной реакции по отношению к Н+/K+-АТФазе париетальных клеток
желудка.
В результате взаимодействия генетических
факторов и факторов окружающей среды формируется воспаление слизистой оболочки желудка
с локализацией в теле и дне с последующей потерей желез — атрофией.
Рассмотрим разные варианты формирования структурных изменений
слизистой оболочки желудка в зависимости от статуса инфицирования H. pylori:
1. АИГ
без инфекции H. pylori («чистый»).
2. Сочетание
АИГ и H. pylori.
3. АИГ в
постэрадикационном периоде H. pylori.
Аутоиммунный
гастрит без
инфекции H. pylori («чистый»)
В
результате реализации факторов риска при этом варианте развития событий
происходит разрушение париетальных клеток специфическими антителами (АТ к ПК). Основной
мишенью АТ к ПК являются α- и β-субъединицы протонного насоса, H+/K+-АТФазы,
уникальной для париетальных клеток [24]. В исследованиях in vitro АТ к ПК
продемонстрировали комплемент-зависимую цитотоксическую активность против
париетальных клеток желудка [25]. Вместе с тем в другом исследовании введение АТ
к ПК из сыворотки крови пациентов с АИГ экспериментальным особям приводило к заметному
уменьшению количества париетальных клеток, но без признаков воспаления
слизистой оболочки желудка. Это подтверждает гипотезу о том, что одного
гуморального компартмента иммунного ответа недостаточно, чтобы вызвать АИГ in
vivo, и несмотря на то, что АТ к ПК и антитела к внутреннему
фактору Касла (АТ к ВФК) считаются серологическими маркерами АИГ,
по-видимому, они не играют ключевой роли в апоптозе париетальных клеток у
человека [26]. Позже было установлено, что часть пациентов с АИГ и вовсе являются
серологически негативными [27].
Основная
роль в патогенезе АИГ отводится аутореактивным цитотоксическим Т-клеткам [28, 29].
Согласно исследованию M. D’Elios et al., H+/K+-АТФаза способна индуцировать ex vivo пролиферацию клонов CD4+ Т-клеток слизистой оболочки желудка,
большинство из которых представляют собой Th1 и продуцируют провоспалительные цитокины,
такие как ФНО-α и интерферон-гамма
(ИФН-γ). Продуцируя провоспалительные цитокины, активированные аутореактивные
Т-клетки усиливают иммунный ответ и способствуют апоптозу париетальных клеток
посредством Fas-Fas-лиганда (FasL) и перфорин-гранзимного механизма.
Последующее ремоделирование тканей, поддерживаемое миофибробластами желудка,
может привести к атрофии слизистой оболочки тела желудка. Кроме того, аутореактивные
Т-клетки, специфичные для H+/K+-АТФазы, также запускают выработку иммуноглобулина
В-клетками [30].
При
изолированном АИГ на ранних стадиях изменения в слизистой оболочке тела желудка
неспецифичны, к ним относится мультифокальная инфильтрация лимфоцитами, плазматическими
клетками, а также тучными клетками и эозинофилами. К проявлениям
гипергастринемии вследствие повреждения кислотопродуцирующих желез на данной
стадии относится гиперплазия париетальных клеток. По мере прогрессирования
иммунного воспаления наблюдается диффузная лимфоплазмоцитарная инфильтрация
собственной пластинки, появление и нарастание выраженности атрофии.
Стереотипный
характер динамики течения АИГ включает неатрофические и атрофические изменения слизистой
оболочки желудка. Исчезновение желез тела желудка сопровождается двумя типами
метапластической трансформации: псевдопилорической (ППМ) и кишечной метаплазией
(КМ) [23]. Как правило, КМ представлена полным фенотипом [30]. ППМ
характеризуется замещением специализированных кислотопродуцирующих желез тела
желудка клетками, напоминающими по морфологии и функциональным признакам
пилорические железы, но в отличие от истинной пилорической метаплазии не
является точным воспроизведением нормальных пилорических желез. Морфологически
ППМ проявляется усилением экспрессии муцина 6 (MUC6) и белка трефоилового фактора 2 (TFF2), а также снижением
экспрессии маркеров париетальных и главных клеток [31, 32]. В 2021 г. Y.Wada et al. опубликовали проспективное исследование, показавшее не
только факт развития ППМ в теле желудка в исходе АИГ, но и давшее основания
предполагать существование некоего метапластического континуума: по мере
прогрессирования атрофии ППМ постепенно сменялась пилорической [33]. ППМ
представляет собой важный промежуточный этап в патогенезе желудочной атрофии,
поскольку ее наличие ассоциировано с повышенным риском развития КМ и дисплазии
эпителия желудка [34]. Ранее считалось, что метаплазии желудочного эпителия
могут быть обусловлены клональным расширением и перепрограммированием отдельных
эпителиальных клеток-предшественников [35]. Однако современные исследования с
использованием методов lineage-tracing продемонстрировали, что развитие ППМ при
АИГ происходит без обязательного клонального расширения отдельных
клеток-предшественников. Вместо этого эпителиальные клетки подвергаются
пластическому неклональному перепрограммированию в ответ на хроническое
воспаление и иммуноопосредованную деструкцию париетальных клеток [36, 37]. Ключевыми
механизмами, лежащими в основе неклональной пластичности эпителия желудка при
АИГ, вероятно, являются изменения в локальном микроокружении и активация
специфических сигнальных каскадов, таких как Notch, BMP и Hedgehog.
Воспалительный микроокружающий фон, сопровождающий АИГ, индуцирует выработку
цитокинов и экспрессию факторов роста, способствующих пластическому
перепрограммированию клеток без селективного клонального отбора [31, 38]. Понимание
неклональной природы ППМ важно для разработки подходов к профилактике и терапии
предраковых состояний желудка.
Развитие
атрофии при АИГ может характеризоваться гиперпластическими полиповидными
изменениями, которые иногда ассоциированы с развитием аденом пилорических желез,
а также обязательным развитием линейной и узелковой (микронодулярной) гиперплазии энтерохромаффинных
клеток [39].
В исследовании G. Pivetta et al. при анализе гастробиоптатов
с последующей идентификацией бактериальной микробиоты с помощью секвенирования 16SрРНК выявлено снижение бактериального разнообразия, а видовой состав бактерий у пациентов с АИГ был
представлен Firmicutes, Proteobacteria и Bacteroidetes, Actinobacteria,
Streptococcus, Prevotella [40]. Вероятно, на фоне гипохлоргидрии за счет ослабления
кислотно-защитного барьера создаются условия для изменения состава микробиоты
желудка с потенциальным чрезмерным ростом бактерий, отличных от H. pylori,
при этом роль подобных изменений в канцерогенезе при АИГ еще предстоит оценить.
Таким образом, реализация данного варианта
(«чистый» АИГ) заключается в первоначальном иммуноопосредованном воспалении
кислотопродуцирующей части слизистой оболочки желудка (т. е. неатрофический АИГ) с последующим формированием атрофии с
характерным топографическим фенотипом (атрофия
тела) и наличием специфических серологических реакций (АТ к ПК, АТ к ВФК).
Гипоацидность и снижение внутреннего фактора приводят к мальабсорбции железа и
витамина B12, вызывая железодефицитную и пернициозную анемию соответственно [24].
Эти состояния представляют собой распространенные клинические проявления АИГ. В
частности, железодефицитная анемия наблюдается примерно в 25–50% случаев [41, 42],
тогда как пернициозная анемия может быть выявлена у 15–25% пациентов с АИГ [41,
43]. Витамин B12 –
незаменимый витамин, имеющий решающее значение для различных физиологических
процессов, таких как развитие эритроцитов, синтез ДНК и функционирование
нервной системы [29].
Помимо дефицита железа и витамина B12, у пациентов с АИГ может наблюдаться дефицит
целого ряда других витаминов и микроэлементов, включая витамин C, витамин D и кальций [44, 45]. Основными
патогенетическими механизмами является либо повышенная деградация, либо сниженная
абсорбция в слизистой оболочке желудка, потенциально обусловленная повышенным
уровнем pH и
избыточным бактериальным ростом [46]. Особую роль в патогенезе АИГ отводят
витамину D, который
обладает иммуномодулирующим действием [47]. Интерес к потенциальным
иммуномодулирующим эффектам витамина D растет с момента открытия рецепторов
витамина D (VDR) в моноцитах, дендритных клетках и активированных Т-клетках [48].
Агонисты VDR препятствуют воспалительным реакциям, вызванным Th1, и
ограничивают последующую дифференциацию Th-клеток в сторону провоспалительного фенотипа
[49]. Витамин D
подавляет провоспалительную активность Th1-клеток и секрецию провоспалительных
цитокинов, таких как интерлейкин-2 (ИЛ-2), ИФН-γ и ФНО-α [50–52]. Активная
форма витамина D также подавляет пролиферацию Т-клеток, экспрессию ИЛ-2 [53] и ИФН-γ
на уровне как микроРНК, так и белка в Т-клетках [54]. Предполагается, что недостаточный
уровень витамина D в организме может ухудшить иммунную регуляцию, потенциально
способствуя дебюту или прогрессированию АИГ [46].
Кроме того, атрофия слизистой оболочки тела
желудка и прогрессирующее снижение секреции кислоты ассоциировано с
физиологической гипергастринемией и гиперплазией энтерохромаффинных
клеток, которая в последующем может трансформироваться в
нейроэндокринные опухоли 1-го типа. Повышенный риск развития нейроэндокринной
неоплазии подтверждает необходимость наблюдения за пациентами с атрофическим
АИГ [55].
Редкая
встречаемость предраковых изменений слизистой оболочки желудка, таких как
неполный тип кишечной метаплазии, может объяснить низкий риск рака желудка у
пациентов с АИГ, поскольку метаплазия, экспрессирующая спазмолитический
полипептид, обычно наблюдаемая при АИГ, не включает клональное
перепрограммирование желудочных желез и может рассматриваться как адаптивное
изменение, а не как истинное предраковое поражение. Однако снижение
кислотопродуцирующей функции желудка в связи с потерей париетальных клеток
сопровождается модуляцией микробиоценоза желудка, стимулируя рост некоторых видов
бактерий, таких как стрептококки, которые могут способствовать развитию
предраковых поражений и рака желудка [56].
Современные
представления о связи между аутоиммунным гастритом и риском развития рака
желудка еще остаются предметом активного обсуждения и исследований. В
частности, в последние годы внимание привлекли данные, свидетельствующие о том,
что т. н. чистый АИГ, т. е. АИГ без сопутствующей инфекции H. pylori и без признаков значительного воспаления другой
этиологии, может сопровождаться низким или даже минимальным риском прогрессии в
рак желудка. Наиболее значимыми в этом контексте являются работы группы
итальянских исследователей под руководством Massimo Rugge, которые показали,
что «чистый» АИГ характеризуется относительно доброкачественным течением и
низкой вероятностью неопластической трансформации слизистой оболочки желудка [57,
58].
Тем не
менее ряд исследований демонстрируют противоположные результаты, выявляя связь
между АИГ и увеличением риска рака желудка [59–61]. Данные об ассоциации АИГ с
увеличением риска развития аденокарциномы желудка также подтверждены и в
метаанализе M. Song et al. [62]. Безусловно, необходимы дальнейшие проспективные
исследования, чтобы определить дополнительные факторы и механизмы связи между АИГ
и риском развития рака желудка.
Сочетание аутоиммунного
гастрита и H. pylori
Эпидемиологические исследования свидетельствуют о значительном
числе пациентов с АИГ, которые перенесли или все еще имеют инфекцию H.
рylori, а у 30% пациентов,
инфицированных H. рylori, могут обнаруживаться АТ к ПК [63].
Однако данные о том, может ли H. pylori инициировать иммунологический
процесс, приводящий к развитию АИГ, все еще требуют подтверждения. A. Amedei et al. установили, что активированные in vivo CD4+
Т-клетки слизистой оболочки пациенток с АИГ и инфицированных H. pylori имеют
перекрестную реактивность как с H+/K+-АТФазой, так и с антигенами H. pylori
[64]. В результате этого открытия авторы
сделали вывод о том, что патогенез АИГ может иметь два различных механизма:
один зависит от «чистого» аутоиммунного процесса, направленного против
париетальных клеток, а другой поддерживается инфекцией H. pylori.
Особый
интерес при изучении вопроса об особенностях течении АИГ у инфицированных H.
pylori лиц
представляет феномен молекулярной мимикрии между антигенами бактерии и
эпитопами Н+/К+-АТФазы собственных париетальных клеток желудка [65, 66].
Молекулярная мимикрия представляет собой способность микроорганизма
«копировать» антигенную структуру макроорганизма, что может привести к
перекрестной активации иммунной системы и аутоагрессии. АИГ, ассоциированный с
инфекцией H. pylori, является одним из ярких примеров молекулярной
мимикрии, при котором активированные иммунные CD4+-Th1-клетки, вырабатываемые
на антигены H. рylori, перекрестно реагируют с эпитопами Н+/К+-АТФазы и
приводят к появлению или усугублению уже имеющегося аутоиммунного воспаления слизистой
оболочки желудка [67]. На сегодняшний день идентифицировано девять белков H.
pylori, каждый из которых содержит эпитоп, схожий с эпитопами Н+/К+-АТФазы
[64], в результате чего париетальные клетки становятся мишенью для
цитотоксических Th1-клеток, что ведет к атрофии слизистой оболочки тела желудка
и, как следствие, к гипохлоргидрии.
При
стимуляции Н+/К+-АТФазы чужеродным патогеном высвобождается большое количество
провоспалительных цитокинов, включая ИФН-γ
и ФНО-α [28]. Их локальная продукция приводит к экспрессии молекул MHC II клетками желудка [68], что позволяет последним
презентировать пептиды аутоантигена Н+/К+-АТФазы Т-клеткам, которые
дифференцируются в эффекторные Th1-клетки. Помимо прочего, ИФН-γ индицирует эпителиальные клетки желудка к
экспрессии ко-стимулирующих молекул (CD80 и CD86) и выработке катепсинов,
которые участвуют в процессинге антигена, и, следовательно, эпителиальные
клетки желудка могут действовать как антигенпрезентирующие клетки [69, 70].
Презентация
бактериальных антигенов эпителиальными или париетальными клетками желудка может
привести к активации H. pylori специфичных FasL Т-клеток желудка, которые уничтожают
антигенпрезентирующие клетки посредством антигензависимых механизмов (перфорин-опосредованный
лизис или индукция апоптоза). Кроме того, презентация желудочной Н+/К+-АТФазы
антиген-презентирующими и эпителиальными клетками желудка специфическим
аутореактивным Т-клеткам может стимулировать дальнейшую активацию Т-клеток и
еще большее увеличение числа Th1-клеток, специфичных для Н+/К+-АТФазы [71, 72].
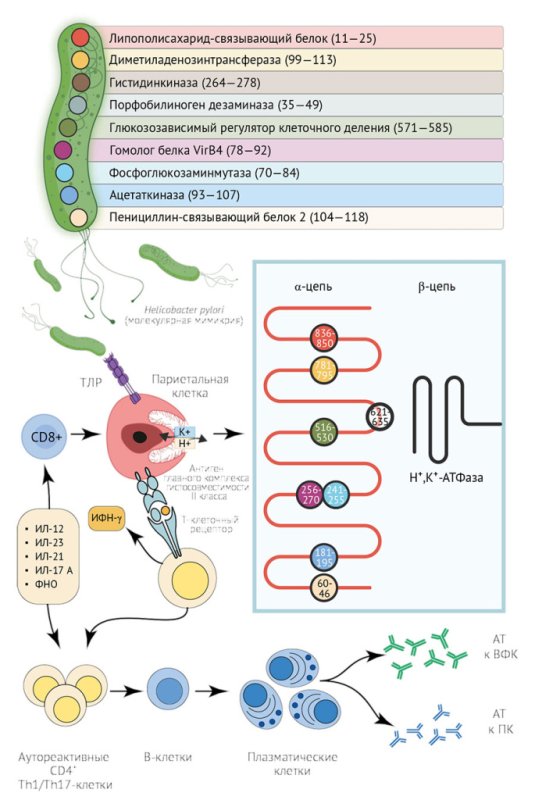
Механизм молекулярной мимикрии между антигенами H. pylori и эпитопами
H+/K+-АТФазой представлен на рисунке [73].
Рисунок. Молекулярная мимикрия между H. pylori и
H+/K+-АТФазой
С практической точки зрения важно отметить, что диагностика инфекционного статуса H. pylori у пациентов с АИГ часто
затруднена. С одной стороны, возможны ложноположительные результаты вследствие
колонизации слизистой оболочки желудка другими уреазообразующими бактериями, с другой
– ложноотрицательные из-за уменьшения количества бактериальных клеток или спонтанного исчезновения
H. pylori при формировании выраженной атрофии [74].
При
сочетании АИГ с инфекцией H. pylori при морфологическом исследовании наблюдаются
типичные для АИГ изменения в теле желудка с вовлечением антрального отдела. H.
pylori-ассоциированный
гастрит характеризуется поверхностным лимфоплазмоцитарным воспалением собственной
пластинки с нейтрофильной инфильтрацией эпителия, в более глубоких слоях
слизистой оболочки могут определяться лимфоидные фолликулы [75]. По мере прогрессирования воспаления при H.
pylori-ассоциированном
гастрите в антральном отделе будут отмечаться очаги атрофии, которые со
временем сливаются, образуя более крупные участки атрофической и метапластической
слизистой оболочки, и в конечном итоге распространяются на тело и дно желудка с
развитием мультифокального гастрита.
Ситуация
сосуществования воспаления и атрофии желез в антральном отделе с атрофией тела
желудка при АИГ является результатом предшествующей или текущей инфекции H.
pylori. Высказано предположение, что антигенная мимикрия между
бактериальными и париетальными клеточными аутоантигенами может этиологически
связывать АИГ с предшествующей или текущей инфекцией H. pylori (т. н. вторичный АИГ) [64].
У
пациентов с АИГ роль шкал Operative Link on
Gastritis Assessment (OLGA) и Operative Link on Gastritic Intestinal Metaplasia Assessment (OLGIM) в прогнозировании эпителиальных неопластических
поражений желудка необходимо изучить дополнительно. Поскольку при первичном АИГ
антрум не затрагивается, стадия OLGA у таких пациентов никогда не превышает II
стадию. Стадии III–IV убедительно свидетельствуют о предшествующей инфекции H.
pylori, которая привела к атрофическим поражениям антрума. У пациентов с
АИГ, имеющих высокую стадию OLGA (связанную с влиянием H. pylori), риск
развития неоплазии желудка оценивается в диапазоне от 6,3 до 25%, поэтому они
нуждаются в динамическом наблюдении [76]. Европейское общество
гастроинтестинальной эндоскопии (ESGE), Европейская группа по изучению
хеликобактера и микробиоты (EHMSG) и Европейское общество патологии (ESP)
рекомендуют, чтобы пациенты с обширными эндоскопическими изменениями (Kimura – Takemoto
C3+ или EGGIM 5+) или поздними гистологическими стадиями атрофического гастрита
(тяжелые атрофические изменения или изменения как в антральном отделе, так и в
теле желудка, OLGA/OLGIM III/IV) проходили высококачественное эндоскопическое исследование
каждые 3 года, независимо от страны происхождения человека [77].
У
пациентов с хроническим гастритом интегральным показателем риска развития рака
желудка является оценка стадии атрофии с использованием шкалы OLGA, а также
стадии кишечной метаплазии с помощью шкалы OLGIM, которые были разработаны в 2005 г.
международными экспертами гастроэнтерологии и патологии [78]. В 2009 г. был
утвержден российский пересмотр системы OLGA,
который значительно упрощает идентификацию атрофии в гастробиоптатах [4,
23]. Однако обращает на себя внимание расхождение в значениях при подсчете
стадии гастрита у пациентов с выраженной атрофией в теле желудка и отсутствием
изменений в антральном отделе. Так, по данным международной системы OLGA, складывается стадия II, а
согласно отечественной редакции – III (табл.).
Таблица. Оценка стадии гастрита на основании международной системы OLGA (А), российский пересмотр международной классификации хронического гастрита (В)
А
Антрум | Тело | |||
Нет атрофии (показатель 0) | Слабая атрофия (показатель 1) | Умеренная атрофия (показатель 2) | Выраженная атрофия (показатель 3) | |
Нет атрофии (показатель 0) | Стадия 0 | Стадия I | Стадия II | Стадия II |
Слабая атрофия (показатель 1) | Стадия I | Стадия I | Стадия II | Стадия III |
Умеренная атрофия (показатель 2) | Стадия II | Стадия II | Стадия III | Стадия IV |
Выраженная атрофия (показатель 3) | Стадия III | Стадия III | Стадия IV | Стадия IV |
В
Антрум | Тело | |||
Нет атрофии (показатель 0) | Слабая атрофия (показатель 1) | Умеренная атрофия (показатель 2) | Выраженная атрофия (показатель 3) | |
Нет атрофии (показатель 0) | Стадия 0 | Стадия I | Стадия II | Стадия III |
Слабая атрофия (показатель 1) | Стадия I | Стадия I | Стадия II | Стадия III |
Умеренная атрофия (показатель 2) | Стадия II | Стадия II | Стадия III | Стадия IV |
Выраженная атрофия (показатель 3) | Стадия III | Стадия III | Стадия IV | Стадия IV |
В
консенсусе RE.GA.IN отмечено, что наличие у пациента с АИГ
стадии III–IV убедительно свидетельствует о предшествующей инфекции H. pylori,
которая явилась причиной атрофических изменений в антральном отделе. Данный
вопрос в отношении подсчета стадии гастрита у пациентов с АИГ и изолированной
выраженной атрофией в теле желудка остается спорным, т. к. полученная
стадия II в данном случае, согласно
международной системе OLGA, не относится к
предраковым изменениям и не требует дальнейшего наблюдения, что сопряжено с
дополнительными рисками у этих пациентов.
Аутоиммунный
гастрит после
эрадикации H. pylori
В многочисленных клинических исследованиях
доказана важная роль эрадикационной терапии как меры канцеропревенции у всех
пациентов с H.
pylori-ассоциированным хроническим
гастритом, однако дискутабельным
остается вопрос относительно влияния эрадикации на последующее течение АИГ.
Появление данных о возможности инфекции H. pylori
индуцировать аутоиммунный процесс в отношении H+/K+-АТФазы с развитием атрофии слизистой
оболочки тела желудка натолкнуло исследователей на мысль о том, что эрадикация бактерии
может показать свою эффективность и в лечении АИГ [63]. В 1998 г.
М. Stolte et al. опубликовали клиническое
наблюдение мужчины 21 года с признаками
активного АИГ слизистой оболочки тела желудка с лимфоцитарной инфильтрацией
собственной пластинки, очагами деструкции желез и гипертрофией сохранившихся
париетальных клеток. Серологическое исследование не выявило АТ к ПК и АТ к ВФК,
однако подтвердило наличие антител к H. pylori иммуноглобулинов класса G (IgG) – 243 ЕД/мл, поэтому пациенту была назначена
эрадикационная терапия. Через 15 мес. после эрадикации титр антител H.
pylori IgG снизился до 11 ЕД/мл, воспалительные инфильтраты с очаговой
деструкцией желез тела и гиперплазией париетальных клеток, характерные для АИГ,
уже не выявлялись. Авторы сделали вывод о том, что АИГ, ассоциированный с персистенцией
H. pylori, возможно вылечить с помощью эрадикации, но необходимы
дальнейшие контролируемые проспективные исследования [79]. Необходимо отметить, что в данном клиническом примере диагноз АИГ был
выставлен на основании эндоскопических и гистологических данных и сообщается об
отсутствии у пациента АТ к ПК и АТ к ВФК.
Позже H. Müller et al. сообщили, что эрадикация H. pylori приводила к исчезновению
активного гастрита у 80% (64 из 80) инфицированных пациентов с неатрофическим
АИГ при динамическом наблюдении со средним периодом до 39,5 мес. [80]. В других
исследованиях сообщалось об обратном развитии атрофии после эрадикации у 20% H.
pylori-позитивных пациентов с атрофическим гастритом тела желудка, и этот
показатель был достоверно выше, чем у H. pylori-негативных пациентов [81,
82].
В 2023
г. Т. Kotera et al. также сообщили о положительном эффекте эрадикации на
течение АИГ, сосуществующего с активной инфекцией H. pylori, у женщины
40 лет. В частности, авторы описали уменьшение выраженности сосудистого рисунка
и исчезновение диффузной гиперемии слизистой оболочки желудка при эндоскопическом
исследовании, снижение титров АТ к ПК (1:40), хотя сохранялись
гистопатологические признаки АИГ (преобладание базальной лимфоцитарной
инфильтрации, деструкция париетальных и главных клеток, псевдопилорическая
метаплазия, гиперплазия энтерохромаффинных клеток) через 7 мес. после
эрадикации, а также дальнейшее улучшение эндоскопической и гистопатологической
картины, снижение титра АТ к ПК (1:20), восстановление париетальных и главных
клеток фундальных желез, нормализация уровня сывороточного гастрина (64 пг/мл) через
26 мес. после эрадикации [83].
Наличие
легкой атрофии (ОШ 2,14; 95% ДИ 1,12–4,1), умеренного воспаления (ОШ 5,3; 95% ДИ
1,64–17,3) и отсутствие кишечной метаплазии (ОШ 2.4; 95% ДИ 1,2–4,8) описаны
как прогностически благоприятные гистопатологические факторы обратного развития
атрофии слизистой оболочки желудка после эрадикации у H. pylori-позитивных
пациентов с АИГ [82].
Вместе
с тем имеются и другие данные. Так, в отчете N. Sumi et al. продемонстрировано быстрое прогрессирование атрофии слизистой
оболочки желудка после эрадикации у H. pylori-позитивного пациента с АИГ [84]. В июне 2023
г. T. Ihara et al. опубликовано клиническое
наблюдение дебюта АИГ с быстрым (в течение 3 лет) прогрессированием
атрофических изменений в теле желудка после эрадикации у пациентки 73 лет с
длительным предшествующим анамнезом H. pylori-ассоциированного гастрита [85].
Несмотря
на имеющуюся разнородность данных всем пациентам
с АИГ, инфицированным H.
pylori, рекомендовано проведение
эрадикационной терапии с последующим динамическим контролем и пристальной
оценкой состояния слизистой оболочки желудка в каждом конкретном случае.
Безусловно, необходимо дальнейшее накопление данных и проведение хорошо спланированных
рандомизированных клинических исследований, которые предоставят
дополнительную информацию о долгосрочном влиянии эффекта эрадикации на течение
АИГ.
Заключение
Согласно
классическим представлениям каскада P. Correa развитие рака желудка можно описать определенной
последовательной сменой патологических изменений. Этот каскад определяет
совершенно четкую последовательность канцерогенеза аденокарциномы желудка: от
неизменной слизистой оболочки к неатрофическому гастриту, затем атрофии, КМ,
дисплазии и в конечном итоге аденокарциноме.
Инфекция
H. pylori и АИГ рассматриваются сегодня в качестве ведущих
этиологических факторов гастрита с высоким риском формирования атрофии и КМ
слизистой оболочки желудка. Рассмотрение молекулярных механизмов развития
атрофии как при изолированном («чистом») АИГ, так и в условиях сопутствующей
либо имевшейся ранее инфекции H. pylori позволяет приблизиться к персонифицированной
оценке риска развития рака. Наряду с риском формирования нейроэндокринной
опухоли на фоне аутоиммунного воспаления слизистой оболочки желудка, риск
аденокарциномы увеличивается при инфицировании H. pylori.
Список литературы / References
Развернуть
- Rugge M, Genta RM, Malfertheiner P, Dinis-Ribeiro M, El-Serag H, Graham DY et al. RE. GA. IN.: the Real-world Gastritis Initiative-updating the updates. Gut. 2024;73(3):407-441. https://doi.org/10.1136/gutjnl-2023-331164.
- Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. https://doi.org/10.1136/bmj.n71.
- Arango MT, Perricone C, Kivity S, Cipriano E, Ceccarelli F, Valesini G, Shoenfeld Y. HLA-DRB1 the notorious gene in the mosaic of autoimmunity. Immunol Res. 2017;65(1):82-98. https://doi.org/10.1007/s12026-016-8817-7.
- Ungar B, Mathews JD, Tait BD, Cowling DC. HLA patterns in pernicious anaemia. Br Med J. 1977;1(6064):798-800. https://doi.org/10.1136/bmj.1.6064.798.
- Ungar B, Mathews JD, Tait BD, Cowling DC. HLA-DR patterns in pernicious anaemia. Br Med J. 1981;282(6266):768-770. https://doi.org/10.1136/bmj.282.6266.768.
- Lahner E, Spoletini M, Buzzetti R, Corleto VD, Vannella L, Petrone A, Annibale B. HLA-DRB1*03 and DRB1*04 are associated with atrophic gastritis in an Italian population. Dig Liver Dis. 2010;42(12):854-859. https://doi.org/10.1016/j.dld.2010.04.011.
- Oksanen AM, Haimila KE, Rautelin HI, Partanen JA. Immunogenetic characteristics of patients with autoimmune gastritis. World J Gastroenterol. 2010;16(3):354-358. https://doi.org/10.3748/wjg.v16.i3.354.
- Pan Y, Wang X, He Y, Lin S, Zhu M, Li Y et al. Tumor suppressor ATP4B serve as a promising biomarker for worsening of gastric atrophy and poor differentiation. Gastric Cancer. 2021;24(2):314-326. https://doi.org/10.1007/s10120-020-01128-7.
- Garelli S, Dalla Costa M, Sabbadin C, Barollo S, Rubin B, Scarpa R, et al. Autoimmune polyendocrine syndrome type 1: an Italian survey on 158 patients. J Endocrinol Invest. 2021;44(11):2493-2510. https://doi.org/10.1007/s40618-021-01585-6.
- Fichna M, Żurawek M, Słomiński B, Sumińska M, Czarnywojtek A, Rozwadowska N et al. Polymorphism in BACH2 gene is a marker of polyglandular autoimmunity. Endocrine. 2021;74(1):72-79. https://doi.org/10.1007/s12020-021-02743-9.
- Calvete O, Reyes J, Valdés-Socin H, Martin P, Marazuela M, Barroso A et al. Alterations in SLC4A2, SLC26A7 and SLC26A9 Drive Acid-Base Imbalance in Gastric Neuroendocrine Tumors and Uncover a Novel Mechanism for a Co-Occurring Polyautoimmune Scenario. Cells. 2021;10(12):3500. https://doi.org/10.3390/cells10123500.
- Laisk T, Lepamets M, Koel M, Abner E; Estonian Biobank Research Team, Mägi R. Genome-wide association study identifies five risk loci for pernicious anemia. Nat Commun. 2021;12(1):3761. https://doi.org/10.1038/s41467-021-24051-6.
- Săsăran MO, Meliț LE, Dobru ED. MicroRNA Modulation of Host Immune Response and Inflammation Triggered by Helicobacter pylori. Int J Mol Sci. 2021;22(3):1406. https://doi.org/10.3390/ijms22031406.
- Jiao Y, Yan Z, Yang A. Mitochondria in innate immunity signaling and its therapeutic implications in autoimmune diseases. Front Immunol. 2023;14:1160035. https://doi.org/10.3389/fimmu.2023.1160035.
- Meliț LE, Mărginean CO, Mărginean CD, Mărginean MO. The Relationship between Toll-like Receptors and Helicobacter pylori-Related Gastropathies: Still a Controversial Topic. J Immunol Res. 2019;2019:8197048. https://doi.org/10.1155/2019/8197048.
- Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9(6):7204-7218. https://doi.org/10.18632/oncotarget.23208.
- Smith SM. Role of Toll-like receptors in Helicobacter pylori infection and immunity. World J Gastrointest Pathophysiol. 2014;5(3):133-146. https://doi.org/10.4291/wjgp.v5.i3.133.
- Rad R, Prinz C, Neu B, Neuhofer M, Zeitner M, Voland P et al. Synergistic effect of Helicobacter pylori virulence factors and interleukin-1 polymorphisms for the development of severe histological changes in the gastric mucosa. J Infect Dis. 2003;188(2):272-281. https://doi.org/10.1086/376458.
- Backert S, Schmidt TP, Harrer A, Wessler S. Exploiting the Gastric Epithelial Barrier: Helicobacter pylori’s Attack on Tight and Adherens Junctions. Curr Top Microbiol Immunol. 2017;400:195-226. https://doi.org/10.1007/978-3-319-50520-6_9.
- Müller A, Solnick JV. Inflammation, immunity, and vaccine development for Helicobacter pylori. Helicobacter. 2011;16(Suppl. 1):26-32. https://doi.org/10.1111/j.1523-5378.2011.00877.x.
- Moulton VR. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front Immunol. 2018;9:2279. https://doi.org/10.3389/fimmu.2018.02279.
- Venter C, Eyerich S, Sarin T, Klatt KC. Nutrition and the Immune System: A Complicated Tango. Nutrients. 2020;12(3):818. https://doi.org/10.3390/nu12030818.
- Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis - pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10(9):529-541. https://doi.org/10.1038/nrgastro.2013.101.
- Lenti MV, Rugge M, Lahner E, Miceli E, Toh BH, Genta RM et al. Autoimmune gastritis. Nat Rev Dis Primers. 2020;6(1):56. https://doi.org/10.1038/s41572-020-0187-8.
- De Aizpurua HJ, Cosgrove LJ, Ungar B, Toh BH. Autoantibodies cytotoxic to gastric parietal cells in serum of patients with pernicious anemia. N Engl J Med. 1983;309(11):625-629. https://doi.org/10.1056/NEJM198309153091102.
- Tanaka N, Glass VB. Effect of prolonged aministration of parietal cell antibodies from patients with atrophic gastritis and pernicious anemia on the parietal cell mass and hydrochloric acid output in rats. Gastroenterology. 1970;58(4):482-494. https://doi.org/10.1016/S0016-5085(70)80062-2.
- Lenti MV, Miceli E, Cococcia S, Klersy C, Staiani M, Guglielmi F et al. Determinants of diagnostic delay in autoimmune atrophic gastritis. Aliment Pharmacol Ther. 2019;50(2):167-175. https://doi.org/10.1111/apt.15317.
- D’Elios MM, Bergman MP, Azzurri A, Amedei A, Benagiano M, De Pont JJ et al. H(+),K(+)-atpase (proton pump) is the target autoantigen of Th1-type cytotoxic T cells in autoimmune gastritis. Gastroenterology. 2001;120(2):377-386. https://doi.org/10.1053/gast.2001.21187.
- Iwamuro M, Tanaka T, Otsuka M. Update in Molecular Aspects and Diagnosis of Autoimmune Gastritis. Curr Issues Mol Biol. 2023;45(7):5263-5275. https://doi.org/10.3390/cimb45070334.
- Zhang T, Tang X. Beyond metaplasia: unraveling the complex pathogenesis of autoimmune atrophic gastritis and its implications for gastric cancer risk. QJM. 2025;118(4):203-247. https://doi.org/10.1093/qjmed/hcaf028.
- Goldenring JR. Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J Pathol. 2018;245(2):132-137. https://doi.org/10.1002/path.5066.
- Weis VG, Sousa JF, LaFleur BJ, Nam KT, Weis JA, Finke PE, et al. Heterogeneity in mouse spasmolytic polypeptide-expressing metaplasia lineages identifies markers of metaplastic progression. Gut. 2013;62(9):1270-1279. https://doi.org/10.1136/gutjnl-2012-302401.
- Wada Y, Nakajima S, Kushima R, Takemura S, Mori N, Hasegawa H et al. Pyloric, pseudopyloric, and spasmolytic polypeptide-expressing metaplasias in autoimmune gastritis: a case series of 22 Japanese patients. Virchows Arch. 2021;479(1):169-178. https://doi.org/10.1007/s00428-021-03033-5.
- Weis VG, Goldenring JR. Current understanding of SPEM and its standing in the preneoplastic process. Gastric Cancer. 2009;12(4):189-197. https://doi.org/10.1007/s10120-009-0527-6.
- McDonald SA, Greaves LC, Gutierrez-Gonzalez L, Rodriguez-Justo M, Deheragoda M, Leedham SJ et al. Mechanisms of field cancerization in the human stomach: the expansion and spread of mutated gastric stem cells. Gastroenterology. 2008;134(2):500-510. https://doi.org/10.1053/j.gastro.2007.11.035.
- Burclaff J, Mills JC. Plasticity of differentiated cells in wound repair and tumorigenesis, part I: stomach and pancreas. Dis Model Mech. 2018;11(7):dmm033373. https://doi.org/10.1242/dmm.033373.
- Willet SG, Mills JC. Stomach Organ and Cell Lineage Differentiation: from Embryogenesis to Adult Homeostasis. Cell Mol Gastroenterol Hepatol. 2016;2(5):546-559. https://doi.org/10.1016/j.jcmgh.2016.05.006.
- Sáenz JB, Mills JC. Acid and the basis for cellular plasticity and reprogramming in gastric repair and cancer. Nat Rev Gastroenterol Hepatol. 2018;15(5):257-273. https://doi.org/10.1038/nrgastro.2018.5.
- Okano A, Takakuwa H, Matsubayashi Y. Parietal-cell hyperplasia mimicking sporadic fundic gland polyps in the atrophic mucosa of autoimmune gastritis. Gastrointest Endosc. 2007;66(2):394-395. https://doi.org/10.1016/j.gie.2007.01.022.
- Pivetta G, Dottori L, Fontana F, Cingolani S, Ligato I, Dilaghi E et al. Gastric Microbiota Gender Differences in Subjects with Healthy Stomachs and Autoimmune Atrophic Gastritis. Microorganisms. 2023;11(8):1938. https://doi.org/10.3390/microorganisms11081938.
- Marignani M, Delle Fave G, Mecarocci S, Bordi C, Angeletti S, D’Ambra G et al. High prevalence of atrophic body gastritis in patients with unexplained microcytic and macrocytic anemia: a prospective screening study. Am J Gastroenterol. 1999;94(3):766-772. https://doi.org/10.1111/j.1572--0241.1999.00949.x.
- De Block CE, Van Campenhout CM, De Leeuw IH, Keenoy BM, Martin M, Van Hoof V et al. Soluble transferrin receptor level: a new marker of iron deficiency anemia, a common manifestation of gastric autoimmunity in type 1 diabetes. Diabetes Care. 2000;23(9):1384-1388. https://doi.org/10.2337/diacare.23.9.1384.
- Esposito G, Dottori L, Pivetta G, Ligato I, Dilaghi E, Lahner E. Pernicious Anemia: The Hematological Presentation of a Multifaceted Disorder Caused by Cobalamin Deficiency. Nutrients. 2022;14(8):1672. https://doi.org/10.3390/nu14081672.
- Aditi A, Graham DY. Vitamin C, gastritis, and gastric disease: a historical review and update. Dig Dis Sci. 2012;57(10):2504-2515. https://doi.org/10.1007/s10620-012-2203-7.
- Cavalcoli F, Zilli A, Conte D, Massironi S. Micronutrient deficiencies in patients with chronic atrophic autoimmune gastritis: A review. World J Gastroenterol. 2017;23(4):563-572. https://doi.org/10.3748/wjg.v23.i4.563.
- Aggeletopoulou I, Konstantakis C, Triantos C. Chronic Atrophic Autoimmune Gastritis: The Evolving Role of Vitamin D. Front Biosci. 2024;29(7):252. https://doi.org/10.31083/j.fbl2907252.
- Johnson CR, Thacher TD. Vitamin D: immune function, inflammation, infections and auto-immunity. Paediatr Int Child Health. 2023;43(4):29-39. https://doi.org/10.1080/20469047.2023.2171759.
- Aranow C. Vitamin D and the immune system. J Investig Med. 2011;59(6):881-886. https://doi.org/10.2310/JIM.0b013e31821b8755.
- Scolletta S, Colletti M, Di Luigi L, Crescioli C. Vitamin D receptor agonists target CXCL10: new therapeutic tools for resolution of inflammation. Mediators Inflamm. 2013;2013:876319. https://doi.org/10.1155/2013/876319.
- Lemire JM, Archer DC, Beck L, Spiegelberg HL. Immunosuppressive actions of 1,25-dihydroxyvitamin D3: preferential inhibition of Th1 functions. J Nutr. 1995;125(Suppl. 6):1704S-1708S. https://doi.org/10.1093/jn/125.suppl_6.1704S.
- Cippitelli M, Santoni A. Vitamin D3: a transcriptional modulator of the interferon-gamma gene. Eur J Immunol. 1998;28(10):3017-3030. https://doi.org/10.1002/(SICI)1521-4141(199810)28:10<3017::AID-IMMU3017>3.0.CO;2-6.
- Alroy I, Towers TL, Freedman LP. Transcriptional repression of the interleukin-2 gene by vitamin D3: direct inhibition of NFATp/AP-1 complex formation by a nuclear hormone receptor. Mol Cell Biol. 1995;15(10):5789-5799. https://doi.org/10.1128/MCB.15.10.5789.
- Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M et al. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol. 2009;183(9):5458-5467. https://doi.org/10.4049/jimmunol.0803217.
- Reichel H, Koeffler HP, Tobler A, Norman AW. 1 alpha,25-Dihydroxyvitamin D3 inhibits gamma-interferon synthesis by normal human peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1987;84(10):3385-3389. https://doi.org/10.1073/pnas.84.10.3385.
- Waldum H, Mjønes P. Towards Understanding of Gastric Cancer Based upon Physiological Role of Gastrin and ECL Cells. Cancers. 2020;12(11):3477. https://doi.org/10.3390/cancers12113477.
- Isakov V. Autoimmune gastritis studies and gastric cancer: True renaissance or bibliometric illusion. World J Gastroenterol. 2024;30(32):3783-3790. https://doi.org/10.3748/wjg.v30.i32.3783.
- Rugge M, Bricca L, Guzzinati S, Sacchi D, Pizzi M, Savarino E et al. Autoimmune gastritis: long-term natural history in naïve Helicobacter pylori-negative patients. Gut. 2023;72(1):30-38. https://doi.org/10.1136/gutjnl-2022-327827.
- Rugge M, Genta RM. Staging and grading of chronic gastritis. Hum Pathol. 2005;36(3):228-233. https://doi.org/10.1016/j.humpath.2004.12.008.
- Kishikawa H, Takarabe S, Ichikawa M, Sasaki A, Nishida J. Gastric Adenocarcinoma in Helicobacter pylori-Negative Autoimmune Gastritis: A Case Report and Literature Review. Cureus. 2024;16(8):e66910. https://doi.org/10.7759/cureus.66910.
- Lahner E, Esposito G, Pilozzi E, Purchiaroni F, Corleto VD, Di Giulio E, Annibale B. Occurrence of gastric cancer and carcinoids in atrophic gastritis during prospective long-term follow up. Scand J Gastroenterol. 2015;50(7):856-865. https://doi.org/10.3109/00365521.2015.1010570.
- Vannella L, Lahner E, Osborn J, Annibale B. Systematic review: gastric cancer incidence in pernicious anaemia. Aliment Pharmacol Ther. 2013;37(4):375-382. https://doi.org/10.1111/apt.12177.
- Faller G, Winter M, Steininger H, Lehn N, Meining A, Bayerdörffer E, Kirchner T. Autoimmune Diseases and Gastric Cancer Risk: A Systematic Review and Meta-Analysis. Cancer Res Treat. 2019;51(3):841-850. https://doi.org/10.4143/crt.2019.151.
- Faller G, Winter M, Steininger H, Lehn N, Meining A, Bayerdörffer E, Kirchner T. Decrease of antigastric autoantibodies in Helicobacter pylori gastritis after cure of infection. Pathol Res Pract. 1999;195(4):243-246. https://doi.org/10.1016/S0344-0338(99)80041-7.
- Amedei A, Bergman MP, Appelmelk BJ, Azzurri A, Benagiano M, Tamburini C, van der Zee R. Molecular mimicry between Helicobacter pylori antigens and H+, K+-adenosine triphosphatase in human gastric autoimmunity. J Exp Med. 2003;198(8):1147-1156. https://doi.org/10.1084/jem.20030530.
- Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2(9):797-801. https://doi.org/10.1038/ni0901-797.
- Wucherpfennig KW. Mechanisms for the induction of autoimmunity by infectious agents. J Clin Invest. 2001;108(8):1097-1104. https://doi.org/10.1172/JCI14235.
- D’Elios MM, Appelmelk BJ, Amedei A, Bergman MP, Del Prete G. Gastric autoimmunity: the role of Helicobacter pylori and molecular mimicry. Trends Mol Med. 2004;10(7):316-323. https://doi.org/10.1016/j.molmed.2004.06.001.
- Valnes K, Huitfeldt HS, Brandtzaeg P. Relation between T cell number and epithelial HLA class II expression quantified by image analysis in normal and inflamed human gastric mucosa. Gut. 1990;31(6):647-652. https://doi.org/10.1136/gut.31.6.647.
- Ye G, Barrera C, Fan X, Gourley WK, Crowe SE, Ernst PB, Reyes VE. Expression of B7-1 and B7-2 costimulatory molecules by human gastric epithelial cells: potential role in CD4+ T cell activation during Helicobacter pylori infection. J Clin Invest. 1997;99(7):1628-1636. https://doi.org/10.1172/JCI119325.
- Barrera C, Ye G, Espejo R, Gunasena S, Almanza R, Leary J et al. Expression of cathepsins B, L, S, and D by gastric epithelial cells implicates them as antigen presenting cells in local immune responses. Hum Immunol. 2001;62(10):1081-1091. https://doi.org/10.1016/s0198-8859(01)00281-6.
- Toh BH, van Driel IR, Gleeson PA. Pernicious anemia. N Engl J Med. 1997;337(20):1441-1448. https://doi.org/10.1056/NEJM199711133372007.
- Mårdh S, Song YH. Characterization of antigenic structures in autoimmune atrophic gastritis with pernicious anaemia. The parietal cell H,K-ATPase and the chief cell pepsinogen are the two major antigens. Acta Physiol Scand. 1989;136(4):581-587. https://doi.org/10.1111/j.1748-1716.1989.tb08705.x.
- Bordin DS, Livzan MA, Mozgovoi SI, Gaus OV. Autoimmune Gastritis and Helicobacter pylori Infection: Molecular Mechanisms of Relationship. Int J Mol Sci. 2025;26:7737. https://doi.org/10.3390/ijms26167737.
- Furuta T, Baba S, Yamade M, Uotani T, Kagami T, Suzuki T et al. High incidence of autoimmune gastritis in patients misdiagnosed with two or more failures of H. pylori eradication. Aliment Pharmacol Ther. 2018;48(3):370-377. https://doi.org/10.1111/apt.14849.
- Choudhuri J, Hall S, Castrodad-Rodriguez CA, Westerhoff M, El Jabbour T, Jain S, Panarelli NC. Features That Aid Identification of Autoimmune Gastritis in a Background of Active Helicobacter pylori Infection. Arch Pathol Lab Med. 2021;145(12):1536-1543. https://doi.org/10.5858/arpa.2020-0615-OA.
- Rugge M, Fassan M, Pizzi M, Zorzetto V, Maddalo G, Realdon S, De Bernard M. Autoimmune gastritis: histology phenotype and OLGA staging. Aliment Pharmacol Ther. 2012;35(12):1460-1466. https://doi.org/10.1111/j.1365-2036.2012.05101.x.
- Dinis-Ribeiro M, Libânio D, Uchima H, Spaander MCW, Bornschein J, Matysiak-Budnik T et al. Management of epithelial precancerous conditions and early neoplasia of the stomach (MAPS III): European Society of Gastrointestinal Endoscopy (ESGE), European Helicobacter and Microbiota Study Group (EHMSG) and European Society of Pathology (ESP) Guideline update 2025. Endoscopy. 2025;57(5):504-554. https://doi.org/10.1055/a-2529-5025.
- Rugge M, Genta RM; OLGA Group. Staging gastritis: an international proposal. Gastroenterology. 2005;129(5):1807-1808. https://doi.org/10.1053/j.gastro.2005.09.056.
- Stolte M, Meier E, Meining A. Cure of autoimmune gastritis by Helicobacter pylori eradication in a 21-year-old male. Z Gastroenterol. 1998;36(8):641-643.
- Müller H, Rappel S, Wündisch T, Bayerdörffer E, Stolte M. Healing of active, non-atrophic autoimmune gastritis by H. pylori eradication. Digestion. 2001;64(1):30-39. https://doi.org/10.1159/000048836.
- Annibale B, Di Giulio E, Caruana P, Lahner E, Capurso G, Bordi C, Delle Fave G. The long-term effects of cure of Helicobacter pylori infection on patients with atrophic body gastritis. Aliment Pharmacol Ther. 2002;16(10):1723-1731. https://doi.org/10.1046/j.1365-2036.2002.01336.x.
- Vannella L, Lahner E, Bordi C, Pilozzi E, Di Giulio E, Corleto VD, et al. Reversal of atrophic body gastritis after H. pylori eradication at long-term follow-up. Dig Liver Dis. 2011;43(4):295-299. https://doi.org/10.1016/j.dld.2010.10.012.
- Kotera T, Nishimi Y, Kushima R, Haruma K. Regression of Autoimmune Gastritis after Eradication of Helicobacter pylori. Case Rep Gastroenterol. 2023;17(1):34-40. https://doi.org/10.1159/000528388.
- Sumi N, Haruma K, Urata N, Tanikawa T, Nakamura J, Suehiro M. Autoimmune gastritis with rapid development of corporal atrophy found after H. pylori eradication therapy, report of a case. I to Chou (Stomach and Intestine). 2019;54(7):1053-1057.
- Ihara T, Ihara N, Kushima R, Haruma K. Rapid Progression of Autoimmune Gastritis after Helicobacter pylori Eradication Therapy. Intern Med. 2023;62(11):1603-1609. https://doi.org/10.2169/internalmedicine.0533-22.
Фото:
Kmpzzz/FOTODOM/Shutterstoсk

 1
1 2
2 3
3 4
4


Комментарии (0)