


Журнал "Медицинский совет" №1/2023
DOI: 10.21518/ms2022-012
Р.А. Гудков1, ORCID: 0000-0002-4060-9692
А.В. Дмитриев1, ORCID: 0000-0002-8202-3876
Н.В. Федина1, ORCID: 0000-0001-6307-7249
В.И. Петрова1, ORCID: 0000-0001-5205-0956
А.Л. Заплатников2, ORCID: 0000-0003-1303-8318
1 Рязанский государственный медицинский университет имени академика И.П. Павлова; 390026, Россия, Рязань, ул. Высоковольтная, д. 9
2 Российская медицинская академия непрерывного профессионального образования; 125993, Россия, Москва, ул. Баррикадная, д. 2/1, стр. 1
Введение. Мукополисахаридоз III типа (синдром Санфилиппо) является редким мультисистемным заболеванием, обусловленным накоплением гликозаминогликанов в клетках различных органов, приводящим к нарушению их функции, специфическим фенотипическим признакам и прогрессирующим нейрокогнитивным нарушениям. Нейронопатические проявления лидируют в клинической картине болезни как по времени манифестации, так и по тяжести. В большинстве случаев на первом и даже втором годах жизни дети имеют нормальное развитие или неотчетливо выраженные отклонения. В отсутствие заместительной ферментотерапии при данном типе мукополисахаридоза быстро развивается грубая задержка интеллектуального и речевого развития, наблюдаются рецидивирующие респираторные эпизоды в виде пневмоний и бронхообструкций, ребенок рано инвалидизируется. Полиморфизм и неспецифичность клинических проявлений, отсутствие настороженности врачей к орфанным заболеваниям являются частой причиной поздней диагностики мукополисахаридоза. В статье представлен обзор данных о распространенности, генетических и фенотипических вариантах мукополисахаридоза III типа, особенностях ведения пациентов и представление клинического наблюдения ребенка с данной патологией.
Клиническое наблюдение. Представлено собственное пролонгированное клиническое наблюдение случая мукополисахаридоза III типа у пациента, находившегося под нашим наблюдением в течение 12 лет. Диагноз установлен и подтвержден в возрасте трех лет. Заболевание манифестировало психоневрологическим регрессом и системными соматическими проявлениями. В динамике у ребенка прогрессировали моторный дефицит, когнитивные нарушения с развитием деменции и рецидивирующий аспирационный синдром.
Заключение. Классическую клиническую картину мукополисахаридоза III типа отличает агрессивное развитие когнитивных и двигательных нарушений на 2–3-м году жизни, характерные фенотипические и соматические проявления болезни. Данный случай демонстрирует множественность проблем и необходимость взаимодействия врачей различных специальностей.
Для цитирования: Гудков Р.А., Дмитриев А.В., Федина Н.В., Петрова В.И., Заплатников А.Л. Мукополисахаридоз III типа: обзор литературы и клиническое наблюдение. Медицинский совет. 2023;17(1):182–188. https://doi.org/10.21518/ms2022-012.
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
Согласие пациентов на публикацию: пациент подписал информированное согласие на публикацию своих данных.
Type III mucopolysaccharidosis: literature review and clinical observation
Roman A. Gudkov1, ORCID: 0000-0002-4060-9692
Andrey V. Dmitriev1, ORCID: 0000-0002-8202-3876
Natalia V. Fedina1, ORCID: 0000-0001-6307-7249
Valeria I. Petrova1, ORCID: 0000-0001-5205-0956
Andrey L. Zaplatnikov2, ORCID: 0000-0003-1303-8318
1 Ryazan State Medical University named after Academician I.P. Pavlov; 9, Vysokovoltnaya St., Ryazan, 390026, Russia
2 Russian Medical Academy of Continuous Professional Education; 2/1, Bldg. 1, Barrikadnaya St., Moscow, 125993, Russia
Introduction. Mucopolysaccharidosis type III (Sanfilippo syndrome) is a rare multi-stem disease caused by the accumulation of glycosaminoglycans (GAG) in the cells of various organs, leading to a violation of their function, specific phentopic signs and progressive neurocognitive disorders. Neurologic manifestations are leading in the clinical picture of the disease, as by the time of manifestation, and by severity. In most patients, in the first and even second years of life, children have normal development or indistinctly pronounced deviations. In the absence of substitute enzyme therapy for this type of MPS, a severe delay in intellectual and speech development develops rapidly, recurrent respiratory episodes in the form of pneumonia and bronchial obstruction are observed, the child is disabled early. Polymorphism and non-specificity of clinical manifestations, lack of alertness of doctors to orphan diseases are a common cause of late diagnosis of MPS. The article presents an overview of data on the prevalence, genetic and phenotypic variants of type III mucopolysaccharidosis, features of patient management and presentation of clinical observation of a child with this pathology.
Clinical observation. We present our own prolonged clinical observation of a type III MPS case in a patient who has been under our supervision for 12 years. The diagnosis was established and confirmed at the age of three years. The disease was manifested by neuropsychiatric regression and systemic somatic manifestations. Motor deficits, cognitive impairments with the development of dementia and recurrent aspiration syndrome progressed in the dynamics of the child.
Conclusions. The classical clinical picture of type III MPS is distinguished by the aggressive development of cognitive and motor disorders at 2–3 years of life, characteristic phenotypic and somatic manifestations of the disease. This case demonstrates the multiplicity of problems and the need for interaction between doctors of various specialties.
For citation: Gudkov R.A., Dmitriev A.V., Fedina N.V., Petrova V.I., Zaplatnikov A.L. Type III mucopolysaccharidosis: literature review and clinical observation. Meditsinskiy Sovet. 2023;17(1):182–188. (In Russ.) https://doi.org/10.21518/ms2022-012.
Conflict of interest: the authors declare no conflict of interest.
Basic patient privacy consent: patient signed informed consent regarding publishing their data.
Введение
Синдром Санфилиппо, или мукополисахаридоз III типа (МПС III), – лизосомальная болезнь накопления с аутосомно-рецессивным типом наследования, вызванная мутациями в генах, кодирующих синтез ферментов, участвующих в расщеплении гепарансульфата, приводящих к его накоплению в лизосомах, и проявляющаяся клинически прежде всего прогрессирующей деменцией, а также различными соматическими нарушениями [1]. Причиной развития болезни являются многочисленные мутации в генах SGSH (17q25.3), NAGLU (17q21.2), HGSNAT (8p11.21) и GNS (12q14), ответственных за выработку четырех различных ферментов и соответствующих четырем подтипам данного МПС: MПС IIIA, B, C и D [2]. Синдром Санфилиппо характеризуется значительным клиническим полиморфизмом по сроку и тяжести поражения нервной системы и соматических проявлений.
Эпидемиология
МПС III типа, вероятно, является наиболее частой формой из всех мукополисахаридозов. По данным систематического обзора Tamas Zelei et al., суммарная частота всех четырех подтипов составляет от 0,17 до 2,35 на 100 000 живорождений при частоте наиболее распространенного МПС IIIA до 1,62 на 100 000 [3]. По другим данным, частота заболевания составляет 0,28–4,1 на 100 000 живорожденных [4, 5]. Общее количество таких пациентов в мире насчитывает 12–19 000 [6]. Распространенность МПС III типа в РФ, по данным Н.В. Бучинской, составляет от 0,88 в Северо-Западном до 0,37 на 100 000 в Центральном федеральных округах.
Отдельные подтипы имеют географическую очаговость, для многих зафиксированных мутаций прослеживается «эффект основателя». Большинство случаев, не связанных с родственными браками, являются сложными гетерозиготами. Расчетная оценка заболеваемости, проведенная на основании анализа частоты носительства патогенных мутаций по данным популяционных геномных данных (Aggregation Consortium Exome и Genome Aggregation Database), определила показатели, превышающие данные региональных регистров пациентов [7]. В особенности это превышение касалось более редких подтипов (C, D), что может быть объяснено наличием стертых и поздних форм. Указанное исследование показало предполагаемую частоту для подтипа А – в Бразилии 0,17–3,27; в Восточной Азии – 0,96–7,56 на 100 000; для подтипа В – в Африке 6,05–9,88; в Южной Азии – 0,08–2,49; для подтипа С – в Африке 0,1–2,36 и для подтипа D – в Африке 0,85–1,72 на 100 000.
Нейронопатические проявления лидируют в клинической картине болезни как по времени манифестации, так и по тяжести. В большинстве случаев на первом и даже втором годах жизни дети имеют нормальное развитие или неотчетливо выраженные отклонения. Начальные проявления болезни – остановка речевого развития и познавательной деятельности – развиваются постепенно. Чаще всего пациенты наблюдаются с диагнозами задержки нервно-психического развития, синдрома дефицита внимания или расстройств аутистического спектра. Синдром Санфилиппо следует исключать у всех пациентов раннего возраста, имеющих проблемы нервно-психического развития, особенно если последние имеют нарастающий характер. Отчетливые нарушения поведения в виде расторможенности, агрессивности, неадекватных эмоциональных реакций, нарушения сна, неспособности к обучению и коммуникации формируются к 3–4 годам. В последующем наблюдается регресс двигательных навыков, бульбарные нарушения, судороги. Вместе с тем у некоторых пациентов возможна относительная сохранность когнитивных функций на втором и даже третьем десятилетии жизни [8].
Несмотря на то что психоневрологические симптомы являются ведущими в клинической картине МПС III типа, именно дисморфические и соматические проявления заболевания могут стать поводом для его диагностики у детей с проблемами нервно-психического развития. Лицевой дисморфизм выражен меньше, чем при других типах МПС, а при подтипах C и D может быть неочевиден. Поражение опорно-двигательного аппарата вариабельно и может включать слабо или умеренно выраженные суставные контрактуры, вертебральные деформации, дисплазию тазобедренных суставов, преждевременную остановку роста, а также специфические изменения в сочленении 1–2-го шейных позвонков (что может представлять проблемы при проведении хирургических и анестезиологических вмешательств). К наиболее демонстративным соматическим проблемам детей с МПС III типа следует отнести гиперплазию лимфоидной ткани глотки с развитием рецидивирующих инфекций лор-органов, хронического отита и тугоухости (как с кондуктивным, так и с нейросенсорным компонентом), обструктивное апноэ сна. Сочетание бульбарных и обструктивных респираторных нарушений способствует развитию аспирационного синдрома, характерны повторные пневмонии и бронхообструктивный синдром. В отличие от других типов МПС для синдрома Санфилиппо в меньшей мере свойственно развитие катаракты, чаще наблюдается поражение сетчатки и зрительного нерва. Гепатомегалия (реже гепатоспленомегалия) также менее выражена и встречается не многим более чем у половины пациентов. Гастроэнтерологические проявления болезни в виде диареи и запоров наблюдаются у большинства пациентов. Кардиопатия с клапанными нарушениями и гипертрофией миокарда редко достигает манифестной выраженности [9–11].
Подтипы МПС III типа клинически проявляют себя различно. Однако в пределах каждого подтипа спектр нейропсихических и соматических синдромов, их тяжесть и возраст манифестации варьируют в широких пределах, что связано с разнообразием мутаций и частотой компаунд-гетерозигот. В наибольшей мере определенные сходные характеристики имеют случаи, описанные в регионально-этнических группах с повышенной заболеваемостью. Подтипы А и В сходны по клинике и характеризуются наиболее агрессивным течением с быстрым развитием деменции и двигательных нарушений и продолжительностью жизни 15–19 лет. По данным корейских исследователей (всего 32 пациента, имевших 7 различных мутаций), у детей с МПС IIIA и IIIB заболевание имело быстропрогрессирующий фенотип с манифестацией в 2,8 ± 0,8 года и постановкой диагноза в 6,3 ± 2,2 года. В качестве наиболее раннего симптома отмечена задержка речи (88,2%), задержка моторики (76,5%), общее отставание (64,7%) и гиперактивность (41,2%). В исследовании A. Meyer et al., включавшем 85 пациентов с МПС IIIA и IIID из Испании и Турции, первые симптомы заболевания дебютировали в 7-месячном возрасте, регрессия речевой, двигательной и когнитивной функций проявилась к 3,3 года с потерей всех трех оцениваемых способностей к 12,5 года [12–14].
Подтипы С и D встречаются существенно реже и имеют большую вариабельность, более позднюю манифестацию и мягкое течение. Возможны атипичные случаи с негрубыми парциальными поведенческими и речевыми расстройствами. Именно подтипы С и D, а также некоторые сложные гетерозиготы подтипов А и В чаще всего имеют позднюю диагностику или остаются недиагностированными [15, 16].
Сообщения о случаях МПС III типа с поздней манифестацией не являются редкостью. Так, D. Lorenz еt al. сообщают о диагностике МПС IIIА типа у пациентки в возрасте 28 лет, сложной гетерозиготе по p.R245H и p.S298P с мягким фенотипом [17]. Интересные сообщения из Нидерландов о 12 взрослых пациентах из 6 семей с MPS III типа (11 MPS IIIA, 1 MPS IIIB) с легким или не нейронопатическим фенотипом, у которых средний возраст постановки диагноза составил 43 года (диапазон 3–68). У восьми пациентов диагноз был установлен семейным скринингом [18].
Эффективного лечения МПС III типа на сегодняшний день нет, основу ведения пациентов составляет симптоматическая терапия, в т. ч. с использованием нейролептиков и антиконвульсантов, а также социально-педагогическое сопровождение [19]. Разрабатываются препараты ферментозаместительной терапии, в т. ч. для интратекального введения [20]. Попытки трансплантации гемопоэтических стволовых клеток не предотвращали неврологическую прогрессию и когнитивные нарушения при относительной стабилизации двигательных способностей [21]. Сообщалось о разработке новой стратегии в лечении с применением аденоассоциированной или лентивирусной векторной опосредованной генной терапии [22].
Приводим собственное пролонгированное клиническое наблюдение ребенка с мукополисахаридозом III типа с 2011 г. до настоящего времени.
Клиническое наблюдение
Впервые ребенок поступил в областную детскую клиническую больницу в возрасте 3 лет с жалобами на субфебрильную температуру, затрудненное носовое дыхание (ночной храп), малопродуктивный приступообразный кашель, одышку. По месту жительства получал антибактериальную терапию без эффекта.
Из анамнеза: родился от III нормально протекавшей беременности, II родов на сроке 35–36 нед., преждевременное излитие околоплодных вод, безводный период 14 ч, слабость родовой деятельности, кесарево сечение. При рождении вес 2 750 г, длина тела 50 см, оценка по шкале Апгар 6/7 баллов. После рождения находился в отделении патологии новорожденных с диагнозом «Перинатальное поражение ЦНС, синдром двигательных расстройств. Врожденная пневмония. Неонатальная желтуха». Грудное вскармливание до 6 мес. В 12 мес. при росте 76 см весил 10 200 г. Держал голову с 2 мес., садился самостоятельно с 8 мес., ходил без опоры с 14 мес. К возрасту 1,5 года речь была представлена несколькими слоговыми словами. Привит по возрасту. Аллергологический анамнез без особенностей. Наследственность не отягощена: родители и сестра здоровы, каких-либо фенотипических особенностей не имеют. Ребенок проживает в отдаленной сельской местности, социально-гигиенические условия и уход удовлетворительные.
С двух лет прослеживается регресс психомоторных навыков: инверсия сна с бодрствованием в ночное время, снизился интерес к совместной игре и эмоциональная окрашенность общения, легко раздражался, при ходьбе появилась хромота. С этого же возраста дебютировали рецидивирующие респираторные эпизоды, выражающиеся нарушением носового дыхания, сопением. На третьем году жизни появились отчетливо выраженные приступы психомоторного возбуждения, сопровождающиеся продолжительным ноющим криком с гримасой, резкими движениями, сопротивлением при попытке успокоить ребенка. Недостаточно реагировал на обращенную речь. Усилились нарушения ночного сна. Общая двигательная активность не нарушалась, но присутствовала гиперактивность, родители испытывали проблемы с контролем за поведением ребенка. В это же время стал поперхиваться жидкой пищей при кормлении. Появились дизурические явления в виде учащения мочеиспусканий, беспокойства, частого упускания мочи, отсутствовал контроль дефекации. Отмечалась постоянная заложенность носа с периодическим появлением слизисто-гнойных выделений и влажным кашлем, а также ночной храп с эпизодами апноэ.
Впервые госпитализирован в инфекционное отделение областной детской клинической больницы в возрасте трех лет по поводу острой респираторной инфекции. При поступлении отмечались умеренно выраженные интоксикационные и респираторные симптомы. Физическое развитие выше среднего +1,9SD, гармоничное, рост 103 см, вес 20 кг. При осмотре обращали на себя внимание фенотипические признаки: грубые черты лица, широкая переносица, короткая шея, эпикант, утолщенный кончик носа, жесткие сухие волосы, темные сросшиеся брови (рис. 1).
Рисунок 1. Пациент М. в возрасте 3 лет (2011 г.)
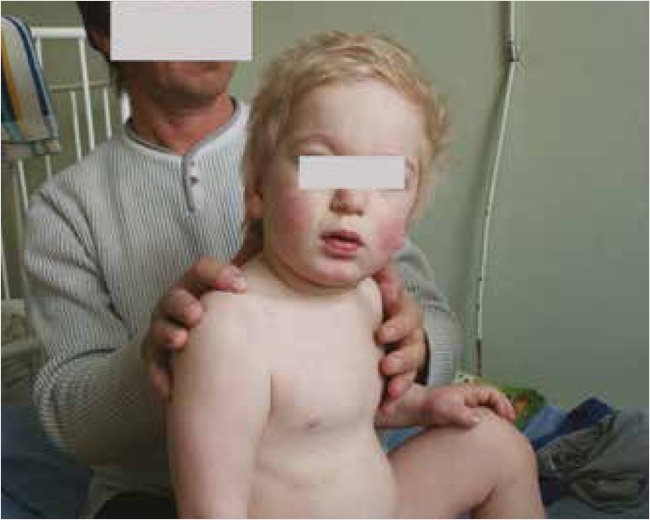
Отмечена умеренно выраженная деформация кистей по типу когтистой лапы с укорочением пальцев. Носовое дыхание затруднено, гипертрофия небных миндалин 2-й степени, рыхлые, отечные, без налетов. Кожа на ощупь плотноватая (утолщенная). Лимфатические узлы не увеличены. Отмечалась одышка с частотой дыхания до 40 в минуту и вспомогательными движениями в плечевом поясе. В легких с обеих сторон выслушивались влажные мелкопузырчатые хрипы. На верхушке сердца при аускультации негромкий систолический шум, тахикардия до 120 в минуту, сатурация 98%. Живот был умеренно увеличен в объеме, определялось увеличение печени и селезенки до 5 и 3 см соответственно с умеренным уплотнением. Отмечались эпизоды жидкого стула.
При осмотре ребенок легко возбудим, отмечена двигательная и речевая расторможенность. Сидел самостоятельно, но при посадке совершал вспомогательные движения. Ходил с поддержкой за руку неуверенно, без поддержки мог упасть. Речь представлена несколькими слоговыми словами, голос низкого тембра, также издавал различные неразборчивые звуки. Обращенную речь родителей понимал и выполнял команды избирательно, контакт ослабевал при нахождении говорящего на расстоянии или вне поля зрения ребенка. В неврологическом статусе отмечена диффузная мышечная гипотония, повышение сухожильных рефлексов. Менингеальных, патологических глазных и очаговых симптомов не выявлялось.
При рентгенологическом исследовании органов грудной клетки визуализировались очаги инфильтрации в верхней доле справа. Эхокардиография сердца выявила перегрузку правых отделов. При УЗИ органов брюшной полости отмечена умеренно выраженная гепатоспленомегалия и гиперплазия мезентериальных лимфатических узлов. Костный возраст соответствовал паспортному.
Фенотипические особенности пациента и динамика психоневрологического статуса позволили заподозрить мукополисахаридоз. Диагноз был подтвержден в лаборатории ФГБНУ «МГНЦ» биохимическим методом по повышенной экскреции с мочой гепарансульфата (107,9 мг/мкмоль креатинина) и дефициту активности гепаран-N-сульфатазы.
В последующем ребенок дважды госпитализировался в отделение психоневрологии и психосоматической патологии ФГАУ «НМИЦ здоровья детей» Минздрава России с диагнозом «Мукополисахаридоз III А типа (синдром Санфилиппо)»; задержка психоречевого развития, псевдобульбарный синдром, вторичная кардиомиопатия (недостаточность митрального, трикуспидального, аортального клапанов и клапана легочной артерии, НК 1-й ст.), множественные контрактуры крупных и мелких суставов конечностей, стеноз шейного отдела позвоночного канала. Терапия включала перициазин (Неулептил) и рисперидон.
В течение периода наблюдения отмечалось медленное прогрессирование симптоматики до полного регресса речи, навыков самообслуживания и самоконтроля. Наибольшие проблемы в уходе за ребенком представляли нарушения ночного сна и приступы психомоторного возбуждения с аутоагрессией в дневное время. Улучшение ночного сна с помощью увеличения дозы перициазина снижало частоту и выраженность дневных приступов. Постепенно нарастали проблемы с приемом пищи: наблюдалось частое поперхивание, кашель при кормлении, попадание жидкой пищи в нос. Во время ночного сна отмечались многократные эпизоды обструктивного апноэ. Не контролировал тазовые функции. Острые респираторные инфекции и аспирационные эпизоды 3–5 раз в год приводили к развитию бронхиальной обструкции с дыхательной недостаточностью. Многократно диагностировался отит.
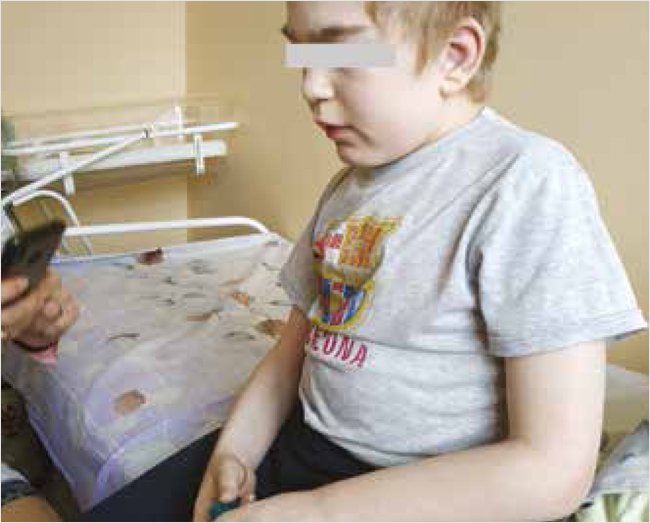
К возрасту 12 лет ребенок имел среднее гармоничное физическое развитие (рост 148 см, вес 38 кг, ИМТ 17,3, окружность головы 60 см). Был неспособен к самообслуживанию, беспомощен, передвигался с поддержкой или в коляске (рис. 2). Отмечалось неадекватное поведение, совершал различные нецеленаправленные и спонтанные движения, в т. ч. самотравмирующие. При попытке осмотра возбуждение нарастало. Узнавал близких и выполнял их простые команды, кратковременно сосредотачивал внимание при общении с родителями. При осмотре внимание ребенка привлекалось кратковременно. Речь в виде немодулированных, негативно окрашенных звуков. Патологическая глазная симптоматика отсутствовала, движение глазных яблок в полном объеме, фотореакция зрачков живая, симметричная. Мышечная сила и тонус снижены. Патологические стопные и кистевые рефлексы, оральные автоматизмы не вызывались. Чувствительность не нарушалась. Функции тазовых органов не контролировал. Отмечалось значительное нарушение носового дыхания, большой язык, гиперплазия десен. Дыхание проводилось по всем полям, жесткое, единичные сухие хрипы с обеих сторон, умеренное тахипноэ до 30–34 в минуту, сатурация 98%. Тоны сердца приглушены, негрубый систолический шум на верхушке и в V точке, частота сердцебиений 90–106 в минуту, артериальное давление 110/75 мм рт. ст. Функция глотания умеренно нарушена, давился пищей и жидкостью. Живот выступающий, край печени пальпировался на 2 см ниже реберной дуги, эластичный, селезенка – край в подреберье. Небольшая пупочная грыжа, умеренно выраженные множественные контрактуры как крупных, так и мелких суставов, асимметрия осанки. Стул один раз в 2–3 дня с тенденцией к запорам.
Рисунок 2. Пациент М. 12 лет (2020 г.)
С целью контроля над усилившимися приступами возбуждения и нарушением ночного сна была предпринята попытка увеличения дозы перициазина, что привело к развитию трехростковой цитопении (тромбоцитопения до 59 × 109/л, лейкопения до 2,7 × 109/л, анемия до 82 г/л), расцененной как реакция на прием нейролептических препаратов. В последующем при снижении дозы препарата отмечена редукция цитопении, но и вместе с тем возвращение тяжести поведенческих нарушений. Прием препарата был продолжен в уменьшенной дозе (9 мг/сут), однако цитопения (с преобладанием тромбоцитопении) сохранялась и периодически нарастала.
В 13 лет ребенок перенес тяжелую полисегментарную пневмонию с плевритом и дыхательной недостаточностью 2-й степени. Усиление дисфагии, неспособность употреблять достаточный объем питания, потеря массы тела и нарушение нутритивного статуса послужили основанием для постановки гастростомы. В послеоперационном периоде назначался трамадол, что сопровождалось уменьшением частоты и выраженности приступов возбуждения. В последующем использование трамадола внутрь в суточной дозе 300 мг было продолжено в связи с положительным эффектом на состояние ребенка и отсутствием других доступных эффективных вариантов.
На амбулаторном этапе паллиативного лечения продолжал получать перициазин, трамадол, каптоприл, верошпирон. За время нахождения дома отмечались эпизоды десатурации после еды и во сне до 88–90%, что требовало дополнительной дотации увлажненного кислорода, по поводу чего ребенок был снабжен кислородным концентратором. В анализах сохранялась выраженная тромбоцитопения. В динамике состояния отмечались прогрессирующие неврологические нарушения, проявившиеся спастичностью и единичным эпизодом клонико-тонических судорог, потребовавшие проведение модификации терапии с назначением габапентина и баклофена.
Заключение
Представленный пациент имеет классическую клиническую картину МПС III типа с агрессивным развитием когнитивных и двигательных нарушений на 2–3-м году жизни, характерными фенотипическими и соматическими проявлениями болезни. Данный случай демонстрирует множественность проблем и необходимость взаимодействия врачей различных специальностей. Мы хотели бы отметить сложность фармакологической коррекции психоневрологической симптоматики и высокий риск побочных действий препаратов и их взаимодействия.
Список литературы / References
- Benetó N., Vilageliu L., Grinberg D., Canals I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int J Mol Sci. 2020;21(21):7819. doi: 10.3390/ijms21217819.
- Бучинская Н.В., Чикова И.А., Исупова Е.А., Калашникова О.В., Костик М.М., Часнык В.Г. Современные подходы к терапии мукополисахаридозов у детей. Вопросы современной педиатрии. 2014;13(3):35–43. doi: 10.15690/vsp.v13i3.1026. / Buchinskaya N.V., Chikova I.А., Isupova E.А., Kalashnikova О.V., Kostik М.М., Chasnyk V.G. Modern approaches to therapy for children with mucopolysaccharidosis. Current Pediatrics. 2014;13(3):35–43. (In Russ.) doi: 10.15690/vsp.v13i3.1026.
- Zelei T., Csetneki K., Vokó Z., Siffel C. Epidemiology of Sanfilippo syndrome: results of a systematic literature review. Orphanet J Rare Dis. 2018;13(1):53. doi: 10.1186/s13023-018-0796-4.
- Buhrman D., Thakkar K., Poe M., Escolar M.L. Natural history of Sanfilippo syndrome type A. J Inherit Metab Dis. 2014;37(3):431–437. doi: 10.1007/s10545-013-9661-8.
- Cross E.M., Grant S., Jones S., Bigger B.W., Wraith J.E., Mahon L.V. et al. An investigation of the middle and late behavioural phenotypes of Mucopolysaccharidosis Type III. J Neurodev Disord. 2014;6(1):46. doi: 10.1186/1866-1955-6-46.
- Kong W., Wu S., Zhang J., Lu C., Ding Y., Meng Y. Global epidemiology of mucopolysaccharidosis type III (Sanfilippo syndrome): an updated systematic review and meta-analysis. J Pediatr Endocrinol Metab. 2021;34(10):1225–1235. doi: 10.1515/jpem-2020-0742.
- Borges P., Pasqualim G., Giugliani R., Vairo F., Matte U. Estimated prevalence of mucopolysaccharidoses from population-based exomes and genomes. Orphanet J Rare Dis. 2020;15(1):324. doi: 10.1186/s13023-020-01608-0.
- Valstar M.J., Ruijter G.J., van Diggelen O.P., Poorthuis B.J., Wijburg F.A. Sanfilippo syndrome: a mini-review. J Inherit Metab Dis. 2008;31(2):240–252. doi: 10.1007/s10545-008-0838-5.
- Wagner V.F., Northrup H. Mucopolysaccharidosis Type III. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W. Amemiya A. (eds.). GeneReviews®. Seattle (WA): University of Washington; 2019. Available at: https://pubmed.ncbi.nlm.nih.gov/.
- Lavery C., Hendriksz C.J., Jones S.A. Mortality in patients with Sanfilippo syndrome. Orphanet J Rare Dis. 2017;12(1):168. doi: 10.1186/s13023-017-0717-y.
- Сарыева О.П., Кулида Л.В., Проценко Е.В., Малышева М.В. Кардиомиопатии у детей – клинические, генетические и морфологические аспекты. Российский медико-биологический вестник им. академика И.П. Павлова. 2020;28(1):99–110. doi: 10.23888/PAVLOVJ202028199-110. / Saryeva O.P., Kulida L.V., Protsenko E.V., Malysheva M.V. Cardiomyopathy in children – clinical, genetic and morphological aspects. I.P. Pavlov Russian Medical Biological Herald. 2020;28(1):99–110. (In Russ.) doi: 10.23888/PAVLOVJ202028199-110.
- Kim M.-S., Yang A., Noh E.-S., Kim C., Bae G.Y., Lim H.H. et al. Natural History and Molecular Characteristics of Korean Patients with Mucopolysaccharidosis Type III. J Pers Med. 2022;12(5):665. doi: 10.3390/jpm12050665.
- Meyer A., Kossow K., Gal A. Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics. 2007;120(5):e1255-e1261. doi: 10.1542/peds.2007-0282.
- Emre S., Terzioglu M., Tokatli A., Coskun T., Ozalp I., Weber B., Hopwood J.J. Sanfilippo syndrome in Turkey: Identification of novel mutations in subtypes A and B. Hum Mutat. 2002;19(2):184–185. doi: 10.1002/humu.9009.
- Ruijter G.J., Valstar M.J., van de Kamp J.M., van der Helm R.M., Durand S., van Diggelen O.P. et al. Clinical and genetic spectrum of Sanfilippo type C (MPS IIIC) disease in The Netherlands. Mol Genet Metab. 2008;93(2):104–111. doi: 10.1016/j.ymgme.2007.09.011.
- Tylki-Szymańska A., Czartoryska B., Górska D., Piesiewicz-Grzonkowska E. Type III D mucopolysaccharidosis (Sanfilippo D): clinical course and symptoms. Acta Paediatr Jpn. 1998;40(5):492–494. doi: 10.1111/j.1442-200x.1998.tb01977.x.
- Lorenz D., Musacchio T., Kunstmann E., Grauer E., Pluta N., Stock A. et al. A case report of Sanfilippo syndrome – the long way to diagnosis. BMC Neurol. 2022;22(1):93. doi: 10.1186/s12883-022-02611-7.
- Nijmeijer S.C.M., van den Born L.I., Kievit A.J.A., Stepien K.M., Langendonk J., Marchal J.P. et al. The attenuated end of the phenotypic spectrum in MPS III: from late-onset stable cognitive impairment to a non-neuronopathic phenotype. Orphanet J Rare Dis. 2019;14(1):249. doi: 10.1186/s13023-019-1232-0.
- Исупова Е.О. Опыт применения арт-терапии в работе с психиатрическими пациентами. Личность в меняющемся мире: здоровье, адаптация, развитие. 2018;6(1):20. Режим доступа: http://humjournal.rzgmu.ru/. / Isupova E.O. The experience of using art therapy in work with psychiatric patients. Personality in a Changing World: Health, Adaptation, Development. 2018;6(1):20. (In Russ.) Available at: http://humjournal.rzgmu.ru/.
- Fedele A.O. Sanfilippo syndrome: causes, consequences, and treatments. ApplClin Genet. 2015;8:269–281. doi: 10.2147/TACG.S57672.
- Köhn A.F., Grigull L., du Moulin M., Kabisch S., Ammer L., Rudolph C., Muschol N.M. Hematopoietic stem cell transplantation in mucopolysaccharidosis type IIIA: A case description and comparison with a genotype-matched control group. Mol Genet Metab Rep. 2020;23:100578. doi: 10.1016/j.ymgmr.2020.100578.
- Seker Yilmaz B., Davison J., Jones S.A., Baruteau J. Novel therapies for mucopolysaccharidosis type III. J Inherit Metab Dis. 2021;44(1):129–147. doi: 10.1002/jimd.12316.

 1
1 2
2 3
3 4
4


Комментарии (0)