


Фото: Chokniti-Studio/FOTODOM/Shutterstoсk
Журнал "Медицинский совет" №18/2025
DOI: 10.21518/ms2025-351
А.А. Корнеенков1, https://orcid.org/0000-0001-5870-8042
Ю.К. Янов2,3, https://orcid.org/0000-0001-9195-128X
Е.Э. Вяземская1, https://orcid.org/0000-0002-4141-2226
А.Ю. Медведева1, https://orcid.org/0009-0002-6921-5299
1 Санкт-Петербургский научно-исследовательский институт уха, горла, носа и речи; 190013, Россия, Санкт-Петербург, ул. Бронницкая, д. 9
2 Военно-медицинская академия имени С.М. Кирова; 194044, Россия, Санкт-Петербург, ул. Академика Лебедева, д. 6
3 Северо-Западный государственный медицинский университет имени И.И. Мечникова; 191015, Россия, Санкт-Петербург, ул. Кирочная, д. 41
Введение. Современный врач вынужден становиться специалистом широкого профиля, сочетая глубокие медицинские знания с техническими компетенциями. Доступность геномных исследований за последние десятилетия резко возросла, однако, для полной их интеграции в медицинскую практику еще существует много препятствий. Учитывая лавинообразный рост новых знаний об ассоциациях геномных данных с болезнями человека, может возникнуть медицинская потребность их самостоятельного анализа для назначения дополнительных медико-генетических исследований, особенно, когда у пациента уже есть полученные ранее NGS-данные (например, экзома).
Цель. Разработать и предоставить детализированное руководство по проведению самостоятельного биоинформатического анализа NGS-данных пациента.
Материалы и методы. Исходными данными являются примеры файлов NGS-данных, предоставляемые пациенту после проведения медико-генетического исследования. Используются реализации как известных, так и самостоятельно разработанных программных алгоритмов выравнивания по референсному геному, обнаружения вариантов, их фильтрации по заданным критериям качества, генам (и их транскриптам) и оценки влияния на здоровье.
Результаты. Разработан общий алгоритм и программный биоинформационный конвейер обработки и анализа данных секвенирования с использованием команд интерфейса Linux, docker-контейнеров известных биоинформатических инструментов bwa, gatk, samtools, bcftools, программ R на основе пакетов проекта Bioconductor и собственных разработок. Этот алгоритм позволяет медицинскому специалисту самостоятельно получать и интерпретировать варианты генетических последовательностей из NGS-данных пациентов.
Выводы. Полученная с помощью этого конвейера информация может служить основой для дальнейших работ по диагностике наследственных заболеваний, персонализированной медицине и фармакогенетике. Использование предложенного алгоритма позволяет достичь поставленных целей и получить на персональных компьютерах варианты геномной последовательности (экзома) пациента, пригодные для последующего анализа и интерпретации. Предполагается, что компьютер врача сможет справиться с подобной задачей за разумное время, обеспечивая надежную и воспроизводимую обработку данных.
Для цитирования: Корнеенков АА, Янов ЮК, Вяземская ЕЭ, Медведева АЮ. От данных секвенирования к пониманию болезни: как врачу обработать NGS-данные пациента на своем компьютере. Медицинский Совет. 2025;(18):108-121. https://doi.org/10.21518/ms2025-351
Конфликт интересов: авторы заявляют об отсутствии конфликта интересов.
From sequencing data to disease understanding: How can a doctor process patient’s NGS data on their own computer
Aleksei A. Korneenkov1, https://orcid.org/0000-0001-5870-8042
Yuri K. Yanov2,3, https://orcid.org/0000-0001-9195-128X
Elena E. Vyazemskaya1, https://orcid.org/0000-0002-4141-2226
Anna Yu. Medvedeva1, https://orcid.org/0009-0002-6921-5299
1 Saint Petersburg Research Institute of Ear, Throat, Nose and Speech; 9, Bronnitskaya St., St Petersburg, 190013, Russia
2 Military Medical Academy named after S.M. Kirov; 6, Akademik Lebedev St., St Petersburg, 194044, Russia
3 North-Western State Medical University named after I.I. Mechnikov; 41, Kirochnaya St., St Petersburg, 191015, Russia
Introduction. In modern medicine, physicians are increasingly required to be versatile specialists, combining in-depth medical knowledge with technical expertise. While the accessibility of genomic research has dramatically increased over the past few decades, its full integration into medical practice still faces significant challenges. Given the rapid proliferation of new knowledge regarding the associations between genomic data and human diseases, there is a growing clinical need for physicians to be able to analyze this data themselves. This is especially true for subsequent medico-genetic studies, particularly when patients already have existing Next-Generation Sequencing (NGS) data (e.g., from exome sequencing).
Aim. The objective of this study is to develop and provide a detailed guide for medical specialists to independently perform bioinformatics analysis of a patient's NGS data.
Materials and methods. The source data for this study are examples of NGS data files provided to patients following a medico-genetic examination. We used both established and custom-developed software algorithms for read alignment against a reference genome, variant discovery, variant filtering based on quality criteria and specific genes (and their transcripts), and assessing their potential health impact.
Results. We developed a comprehensive algorithm and a bioinformatics processing pipeline for sequencing data analysis. This pipeline utilizes a Linux command-line interface, along with Docker containers for established bioinformatics tools such as bwa, gatk, samtools, and bcftools, as well as R scripts based on the Bioconductor project and our own proprietary developments. This algorithm allows medical professionals to independently obtain and interpret genetic variants from a patient's NGS data.
Conclusion. The information obtained through this pipeline can serve as a foundation for further work in diagnosing hereditary diseases, personalized medicine, and pharmacogenetics. The proposed algorithm effectively achieves the study's objective, enabling the retrieval of patient genomic sequence variants (exomes) suitable for subsequent analysis and interpretation on a personal computer. We anticipate that a physician’s computer can handle this task in a reasonable amount of time, ensuring reliable and reproducible data processing.
For citation: Korneenkov AA, Yanov YK, Vyazemskaya EE, Medvedeva AY. From sequencing data to disease understanding: How can a doctor process patient’s NGS data on their own computer. Meditsinskiy sovet = Medical Council. 2025;(18):108-121. (In Russ.) https://doi.org/10.21518/ms2025-351
Conflict of interest: the authors declare no conflict of interest.
Введение
Современный врач
вынужден становиться специалистом широкого профиля, сочетая глубокие
медицинские знания с техническими компетенциями. Интеграция различных
специальностей в медицине стремительно набирает обороты, и игнорировать этот
тренд невозможно [1]. В любых концепциях современной медицины – персонализированная
медицина [2], прецизионная (точная) медицина [3, 4], геномная медицина [5] – фундаментальную
основу составляют геномные исследования, биоинформатика [6] и передовые
инженерные технологии [7, 8].
Ранее врачи могли лишь
наблюдать развитие болезни, теперь же новейшие технологии позволяют вмешиваться
непосредственно в геном: проводить коррекции генных мутаций и эпигенетические
модификации [9]. Например, в области оториноларингологии ранние симптомы потери
слуха нередко являются маркерами серьезных генетических заболеваний,
проявляющихся как сразу, так и позднее в зрелом возрасте [10]. Знание прогноза помогает предупреждать развитие болезней. Современные методы
требуют развития новых подходов к лечению, направленных на устранение
первопричин патологий, включая разработку целенаправленной этиологической и
патогенетической терапии [2].
Доступность геномных
исследований за последние десятилетия резко возросла благодаря развитию
технологий Next Generation Sequencing (NGS) [11], однако на пути полной
интеграции геномных исследований в повседневную клиническую работу до сих пор
существуют препятствия [12]. К ним относятся все еще высокая стоимость
процедур, сложность интерпретации генетических данных, недостаточный уровень
знаний медиков в области биоинформатики, геномики [2] и понимания ими
их практической ценности [13].
В российских
университетах наблюдается постепенное включение элементов биоинформатики в
учебные планы медицинских вузов. Некоторые медицинские академии, университеты
вводят курсы, посвященные основам биоинформатики, молекулярной биологии, генной
инженерии и цифровым технологиям. Однако такая практика пока носит скорее
экспериментальный характер и не охватывает все учебные заведения. Во многих
странах мира биоинформатика стала неотъемлемой частью современного медицинского
образования [14–16]. По биоинформатике вводятся обязательные образовательные
модули, начиная с бакалавриата и продолжая в магистратуре и докторантуре [17–19].
К примеру, в США многие американские медицинские школы предлагают
специализированные курсы по биоинформатике и вычислительной биологии [20, 21].
Студенты имеют возможность изучать принципы анализа геномных данных, машинного
обучения и статистического моделирования. Европейские университеты, такие как
Оксфорд, Кембридж и Эдинбургский университет, давно ввели образовательные
программы, включающие биоинформатику и вычислительные методы анализа данных [22].
Несмотря на осознание потребности и растущую важность биоинформатики в
современной медицине, ее полноценное внедрение в образовательный процесс еще впереди
[14, 23, 24].
Интерес врачей к
геномным исследованиям возрастет, если они поймут, что знания в области
биоинформатики и доступ к NGS- данным позволяют не только подтверждать выводы
медицинских экспертов, но и самостоятельно проводить глубокие аналитические
исследования для понятных практических задач, многократно используя один и тот
же набор геномных данных пациента [25].
Подобные исследования
доступны врачам любого уровня подготовки благодаря наличию открытых программных
решений (bwa, samtools, gatk, R [26], Python и др.), позволяющих организовать
биоинформатические конвейеры (сценарии) и проводить комплексный анализ на
современном офисном компьютере. Эффективность работы с этими инструментами
повышается благодаря современным технологиям контейнеризации [27], таким как
Docker, облегчающим запуск готовых, предварительно настроенных образов
программных продуктов и алгоритмов, устраняя необходимость сложной
самостоятельной настройки программного обеспечения. Также способствует успеху
наличие у врача навыков работы в операционных системах на базе Linux,
получивших широкое распространение в медицинских учреждениях, образовательных
центрах и исследовательских лабораториях. Однако вовсе не обязательно
кардинально менять привычную рабочую среду (Windows или macOS) ради освоения
Linux. Существует ряд технологий, позволяющих использовать преимущества Linux
параллельно с существующими рабочими условиями: виртуализация,
Docker-контейнеры, средства Windows для работы с Linux-программами, а также
удаленная работа с мощными Linux-серверами.
Благодаря искусственному
интеллекту и специализированным помощникам стало проще осваивать цифровые
технологии, Linux и языки программирования [28], таким как российский чат-бот
GigaChat, который способен подсказывать команды, разъяснять алгоритмы и
помогать строить собственные программные конвейеры.
Основной целью настоящей работы является
предоставление детализированного руководства по проведению биоинформатического
анализа данных NGS-секвенирования пациента. Предполагается, что даже личный
компьютер врача сможет справиться с подобной задачей за разумное время,
обеспечивая надежную и воспроизводимую обработку данных. Для демонстрации
процесса в статье приводится набор базовых команд интерфейса командной строки,
доступных в разных операционных системах, поддерживающих Docker (Linux, macOS,
Windows через WSL или Docker Desktop) и сценарии обработки и анализа данных на
языке R [29–34].
Предлагаемые алгоритмы
и их программные реализации могут использоваться врачами для образовательных
целей, научных исследований и практической работы, в ситуациях дефицита или
отсутствия доступа к услугам генетических лабораторий. Особенно, когда на руках
у пациента уже есть полученные ранее NGS-данные (например,
экзома). Это дает возможность своевременно назначать дополнительные
диагностические мероприятия по лабораторной генетике у сертифицированных
специалистов.
Материалы
и методы
Файлы исходных данных
Для биоинформатического
анализа NGS-данных пациента необходимы: (1) результаты секвенирования генома
пациента в виде одного или нескольких файлов формата FASTQ; (2) референсный
геном.
(1) Перед анализом результаты секвенирования должны проходить
предварительную подготовку по объединению, обезличиванию (при необходимости) и
сокращению объема файлов (для учебных примеров). В этой работе использовались
NGS-данные, которые предоставлялись пациенту в медико-генетической лаборатории
в виде файлов многотомного архива с
расширением «.fq.gz» или «.fastq.gz», например, S0001_L00_R1.fq.gz, S0001_L00_R2.fq.gz и
так далее. Эти файлы необходимо объединить (англ., merge) с помощью команды «zcat» в один большой файл, например, S0001_merged_LO.fastq:
zcat
S0001_L0*_R*.fq.gz > S0001_merged_LO.fastq
Деперсонификация данных
NGS-секвенирования, включающая удаление персональной информации или замещение
ее сгенерированными псевдонимами, позволяет обеспечить безопасность
исследований и опубликовывать их результаты. В этом исследовании создается деперсонифицированная
копия исходного файла S0001_merged_LO.fastq
с новым, вручную созданным идентификатором пациента «PID25001».
Ниже представлены
команды оболочки Linux, которые можно запускать последовательно или в виде скрипта
(табл. 1). Первой командой (строка 1) происходит создание
идентификатора пациента (можно выбрать любую удобную форму). Затем требуется
указать название исходного fastq-файла (строка
2) и схему, по которой формируется название нового файла, включающим идентификатор
пациента (строка 3). Далее
производится замена оригинальной информации заголовка fastq-файла на заданный
идентификатор (строка 4),
выводится сообщение о создании нового файла (строка 5). Для того чтобы сопоставить информацию заголовков
исходного и деперсонифицированного файла выполняются команды в строках 6 и 7.
Таблица 1. Команды оболочки Linux для деперсонификации .fastq-файлов
№ | Команда |
1 | IDENTIFIER="PID25001" |
2 | ORIGINAL_FILE="S0001_merged_LO.fastq" |
3 | NEW_FILE="${IDENTIFIER}.fastq" |
4 | sed -E 's/_([[:digit:]]{9})/_000000001/g' "$ORIGINAL_FILE" | sed -E 's/_([[:alnum:]]{21})_/_XXXXXXXXX_/g' > "$NEW_FILE" |
5 | echo "Новый файл успешно создан: $NEW_FILE" |
6 | grep '^@' "$ORIGINAL_FILE" | head -n 1 |
7 | grep '^@' "$NEW_FILE" | head -n 1 |
Теперь файл PID25001.fastq
с деперсифицированными NGS-данными может быть при необходимости переименован и
использован в публикуемых результатах исследования. Однако для образовательных
целей этой статьи такой файл может быть неудобен из-за его размера (в
разархивированном виде, например, экзом, занимает около 30 Гб). Простой
способ сделать небольшой учебный файл необходимого размера – использовать
команду «head».
Эмпирически известно, что около 1 млн прочтений обычно занимает порядка
100–200 Мб, что можно использовать как ориентир для расчета нужного размера
учебного файла.
В
результате выполнения команды head -n 1000000 PID25001.fastq > subset1M_PID25001.fastq получается
файл размером около 100 Мб. При необходимости его можно упаковать в архив с
помощью команды «pigz -k subset1M_PID25001.fastq» («pigz»
удобен тем, что поддерживает многопоточность и ускоряет процесс упаковки /распаковки,
задействуя все доступные ядра процессора).
Таким образом, получив файлы NGS-данных одного пациента (обычно
представляющие собой набор файлов), мы предварительно объединяем их в единый
большой файл и деперсонифицируем его, присваивая условное название, например, PID25001.fastq.
Для возможности воспроизведения предложенного сценария анализа (при
этом сохраняя лишь общую структуру и порядок действий, а не итоговые результаты)
на рис. 1 представлена ссылка [1] на репозиторий BioVarExplorer
на портале GitHub, где можно скачать существенно сокращенный вариант
файла размером примерно 1 млн записей.
Готовые исходные данные (subset1M_PID25001.fastq.gz)
и программные скрипты (VCFtoVarGnomAD.R) для
воспроизведения данного исследования доступны в публичном репозитории
BioVarExplorer на портале GitHub.
Рисунок 1. QR-код на репозиторий BioVarExplorer

При выполнении всех дальнейших инструкций и команд подразумевается
работа именно с файлом полного размера PID25001.fastq,
поэтому везде в примерах команду PID25001.fastq
следует заменять на полный путь к собственному файлу с данными.
(2) Файл референсного генома в формате fasta может
быть скачен с сайта Национального центра биотехнологической информации
(National Center for Biotechnology Information) NCBI [2]. Внутри
этого каталога референсный геном, как правило, в двух файлах от разных
источников, называемых по-разному: от NCBI Refseq – GCF_000001405.40_GRCh38.p14_genomic.fna
и от GenBank – GCA_000001405.29_GRCh38.p14_genomic.fna.
Расширение «.fna»
используется для обозначения файлов, содержащих последовательности нуклеотидов
(ДНК или РНК) в формате FASTA. Термин «.fna» расшифровывается как FASTA
nucleotide sequence.
В качестве абстрактного примера списка генов, по
геномным диапазонам которых производится вывод и аннотация вариантов, в
используемом сценарии на языке R используются следующие гены и их транскрипты: SLC3A1, TTN, GUF1, TMEM70, CTU2 и MKKS.
Готовые исходные данные и программные скрипты для
воспроизведения данного исследования (статьи) доступны в публичном репозитории
BioVarExplorer на портале GitHub [3] .
Программное обеспечение
Настройку программной среды можно максимально
упростить, используя docker-контейнеры. Docker – это технология, которая
позволяет запускать приложения, например, с биоинформатическими инструментами в
изолированном окружении, называемом контейнером [4]. Внутри
контейнеров используется интерфейс командной строки операционной системы linux,
через который осуществляется ввод команд. Этот интерфейс позволяет создавать
сценарии, конвейеры (пайплайны, от англ., pipeline) из различных команд,
понятных операционной системе. Для установки Docker в Windows необходимо
скачать и установить установочный файл с расширением exe. В Linux-системах,
таких как Ubuntu, установка Docker осуществляется через официальные репозитории
или сторонние хранилища. Наиболее распространенный способ установки – использование
менеджера пакетов apt-get (для Debian-based систем, таких как Ubuntu, Mint и
других).
Для выравнивания NGS-данных пациента по
референсному геному и получения файлов с обнаруженными вариантами в формате VCF
удобно использовать уже настроенные Docker-образы: staphb/bwa и broadinstitute/gatk.
staphb/bwa – это Docker-образ, созданный проектом Staphylococcus
Toolkit, который представляет собой сборник инструментов для анализа геномов
бактерий. broadinstitute/gatk – это официальный Docker-образ, предоставляемый
известным Институтом Брода (Broad Institute), который содержит самую свежую
версию инструмента GATK (Genome Analysis Toolkit).
При изложении материалов этой статьи будет
использоваться следующая принятая терминология:
1) Хост-машина (или просто хост) – компьютер, на котором
запущен контейнер Docker.
2) Докер-образ (Docker-образ) – это «упаковка»
приложения вместе с его зависимостями и настройками среды. Докер-образ похож на
шаблон, по которому создаются контейнеры.
3) Контейнер (Container) – это экземпляр образа,
работающий на хост-машине. Сначала загружается докер-образ, а затем запускается
контейнер на основе этого образа.
Для настройки рабочей среды необходимо выполнить
несколько команд, приведенных в табл. 2.
Блок-схема этого процесса приведена на рис.
2. Эти команды могут быть сгруппированы следующим образом: I часть (команд)
– подготовка рабочей среды; II часть – загрузка докер-образов и запуск контейнеров
на их основе; III часть – работа с контейнером от Bioconductor; IV часть –
выполнения основных операций с файлами внутри контейнеров; V часть – завершение
работы с контейнерами. Ниже приводится краткое описание этих команд.
Рисунок 2.
Блок-схема конвейера биоинформатических инструментов: подготовка среды Docker и
загрузка исходных NGS-данных
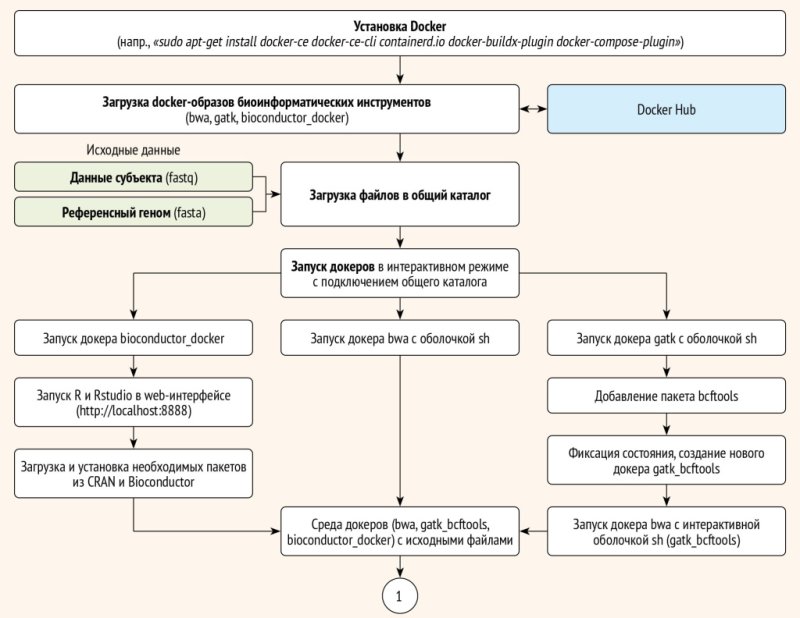
Таблица 2. Перечень команд загрузки, запуска и настройки рабочей среды
№ | Команда |
1 | sudo usermod -aG docker "$USER" |
2 | newgrp docker |
3 | sudo mkdir -p bind && sudo chown "$USER" ~/bind |
4 | docker pull staphb/bwa |
5 | docker pull broadinstitute/gatk |
6 | docker pull bioconductor/bioconductor_docker |
7 | docker run -it --rm -v "$(pwd)/bind":/data --name bwa_ubuntu staphb/bwa:latest sh |
8 | docker run -it --rm -v "$(pwd)/bind":/data --name gatk_ubuntu broadinstitute/gatk:latest sh |
9 | apt-get update && apt-get install -y bcftools |
10 | docker ps |
11 | docker commit -p 59d1f8ee2b89 gatk/bcftools:ver1 |
12 | docker run -it --rm -v "$(pwd)/bind":/data --name gatk_bcftools gatk/bcftools:ver1 sh |
13 | docker run -e PASSWORD=bioc -p 8888:8787 --mount type=bind,source="$(pwd)"/bind,destination=/home/rstudio/R/bind --name bioconductor_vcf bioconductor/bioconductor_docker:latest |
14 | BiocManager::install(c("Biostrings", "GenomicFeatures", "VariantAnnotation", "org.Hs.eg.db", "Bsgenome.Hsapiens.UCSC.hg38", "TxDb.Hsapiens.UCSC.hg38.knownGene", "SNPlocs.Hsapiens.dbSNP155.GRCh38")) |
15 | docker stop bwa_ubuntu |
16 | docker stop gatk_bcftools |
17 | docker stop bioconductor_vcf |
I часть. Подготовка рабочей среды
Правильная настройка прав и разрешений пользователей в Linux
критически важна. Команда в строке 1 табл.
2 добавляет текущего пользователя в группу docker, а перезапуск сессии (строка 2) применяет эти изменения. Для
обмена файлами между хост-машиной и контейнерами необходимо также создать на
хост-машине директорию bind и
назначить ее владельцем текущего пользователя (строка 3).
II
часть. Загрузка Docker-образов и запуск контейнеров с биоинформатическими
инструментами
Для начала требуется загрузить все контейнеры с
использованием терминала операционной системы Linux. Скачивание Docker-образов
bwa, gatk, bioconductor_docker, выполняется с помощью docker pull (строки 4–6). Командой docker run -it --rm -v создается контейнер в интерактивном режиме с
подключением тома (папки) (строки 7–8). В командах в строках 7, 8, 12 параметр sh означает запуск специальной командной
оболочки sh. Переключаясь между
окнами терминалов с оболочкой sh, допустимо
работать сразу в нескольких запущенных контейнерах Docker одновременно, Выход
из оболочки sh производится клавишами
Ctrl + D или вводом команды exit.
Иногда требуется добавить новые пакеты в имеющийся Docker-образ, сохранить образ
под новым именем и использовать в дальнейшем. Например, для добавления пакета bcftools в Docker-образ broadinstitute/gatk:latest и
сохранения под именем gatk/bcftools:ver1
выполняется ряд простых команд:
1) сначала этот пакет устанавливается в запущенный
контейнер gatk (строка 9);
2) в новом окне терминала выводится идентификатор
CONTAINER ID этого контейнера (строка 10);
3) по этому идентификатору измененный контейнер сохраняется
(в приведенном коде 59d1f8ee2b89
нужно изменить на свой) (строка 11).
Теперь на основе Docker-образа gatk/bcftools:ver можно запустить новый контейнер с именем gatk_bcftools (строка 12).
Отдельно необходимо сказать о совместном
использовании общей директории bind хост-машины
всеми запущенными контейнерами. В строке запуска контейнера (например, строка 7)
параметр $(pwd) означает путь к
каталогу на хост-машине (т.е. папке bind), а /data – точка монтирования внутри контейнера. В новом (втором) окне
терминала второй контейнер запускается аналогичным способом (строка 8). Оба
контейнера видят содержимое каталога bind
хост-машины в одной и той же папке /data.
Если создать файл в одном контейнере в папке /data, он автоматически появится в папке /data другого контейнера и в папке bind хост-машины. Контейнер с bcftools (строка 12) также имеет
доступ к общей папке bind через свою
папку /data.
Команды docker images, docker ps –a, docker ps позволяют
узнать какие образы загружены, какие контейнеры запущены и каков их перечень.
III
часть. Настройка работы R/Rstudio в докере bioconductor/bioconductor_docker
Команда в строке 13 инициирует запуск нового
контейнера с именем bioconductor_vcf
на основе Docker-образа bioconductor/bioconductor_docker.
Она передает значение пароля (PASSWORD) bioc
для установки пароля пользователя в приложении, работающем внутри контейнера.
Параметр «-p» указывает, что порт хост-машины (8888) будет перенаправлен на
порт контейнера (8787), что позволяет взаимодействовать с сервисом, запущенном
внутри контейнера, через браузер. Содержимое каталога /bind на хост-машине станет доступным в контейнере по адресу /home/rstudio/R/bind.
Для работы в среде R/RStudio этого контейнера
необходимо открыть браузер и перейти по адресу http://localhost:8888. Логин и
пароль по умолчанию: username – rstudio, password – bioc (он указан в команде
запуска контейнера). Внутри RStudio необходимо установить обязательные
биологические пакеты, используя команду строки 14.
Описание установки и настройки докера проекта
Bioconductor представлено на официальном сайте bioconductor.org [5], там же и
различные варианты настройки рабочей среды. По умолчанию в этом докере уже
установлены многие пакеты для работы с генетическими данными, однако для данных
задач необходимо установить в R дополнительные пакеты геномных аннотаций.
IV
часть. Основные
операции с файлами внутри контейнеров
Чтобы работать с пользовательскими данными внутри докера,
требуется скопировать в папку bind на
хост-машине следующие файлы:
1) полученные из медико-генетической лаборатории
(при необходимости — деперсонифицированные файлы),
2) последовательности референсного генома:
а) PID25001.fastq
б) GCF_000001405.40_GRCh38.p14_genomic.fna.
V
часть. Завершение
работы с контейнерами
Для остановки активных контейнеров используется команда,
состоящая из docker stop и имени
контейнера (строки 15–17).
Таким образом, была проведена основная
подготовительная работа: загружены и установлены docker-контейнеры, настроена
среда R для работы с файлами обнаруженных или «вызванных» вариантов, загружены
файлы результатов секвенирования субъекта исследования (пациента), референсный
геном.
Результаты
После запуска docker-контейнеров (если контейнеры
запущены, они отражаются в списке по команде docker ps) можно приступать к получению и анализу вызванных
вариантов последовательности. Последовательность действий по вызову вариантов
представлена на блок-схеме (рис. 3).
Перед выполнением команд требуется проверить, все ли необходимые файлы доступны
для инструментов и находятся в нужных папках. Последовательность команд
оболочки Linux представлена в табл. 3.
В тексте приводятся ссылки на строки этого кода и краткое описание ключевых
команд: выравнивания – bwa mem, вызова
вариантов – gatk HaplotypeCaller и
фильтрации вариантов – bcftools filter.
Рисунок 3. Блок-схема конвейера
биоинформатических инструментов: выравнивание в docker-среде bwa и вызов
вариантов docker-среде gatk и фильтрация вариантов по критериям качества в
docker-среде bcftools
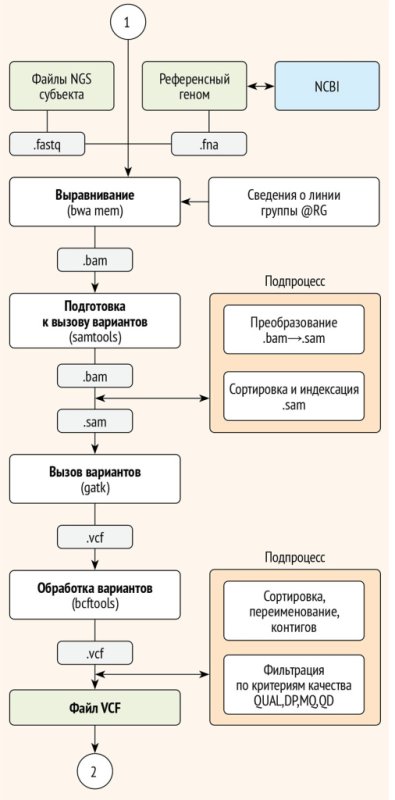
Таблица 3. Перечень
команд оболочки Linux для выравнивания, вызова вариантов и их фильтрации по
выбранным показателям качества
№ | Команда |
1 | bwa index GCF_000001405.40_GRCh38.p14_genomic.fna |
2 | bwa mem -t 8 -R '@RGID:SMP001LB:LBR001PL:NovaSeq6000PU:NS5001234SM:Human-SampleA' GCF_000001405.40_GRCh38.p14_genomic.fna PID25001.fastq > output_PID25001.sam |
3 | samtools view -Sb output_PID25001.sam > output_PID25001.bam |
4 | samtools sort output_PID25001.bam -o sorted_PID25001.bam |
5 | samtools index sorted_PID25001.bam |
6 | samtools view -H sorted_PID25001.bam |
7 | samtools faidx GCF_000001405.40_GRCh38.p14_genomic.fna |
8 | samtools dict GCF_000001405.40_GRCh38.p14_genomic.fna > GCF_000001405.40_GRCh38.p14_genomic.dict |
9 | gatk HaplotypeCaller -R GCF_000001405.40_GRCh38.p14_genomic.fna -I sorted_PID25001.bam -O variants_PID25001.vcf --native-pair-hmm-threads 8 |
10 | bcftools query -f '%CHROM' variants_PID25001.vcf | sort | uniq |
11 | bcftools annotate --rename-chrs rename_NCToChr.txt variants_PID25001.vcf > renamed_variants_PID25001.vcf |
12 | bcftools filter -s LowQual -S . -e 'QUAL < 30 || FORMAT/DP < 20 || MQ < 40 || QD < 2' renamed_variants_PID25001.vcf > filt_PID25001_FORMAT-DP.vcf |
13 | bcftools view -f PASS filt_PID25001_FORMAT-DP.vcf > fin_filt_FDP_PID25001.vcf |
Примечание. Для переноса используется символ
экрана «» без пробела после него.
Представленный код позволяет решить поставленную
задачу обнаружения и фильтрации вариантов в геномной последовательности
пациента и, как в случае подготовки рабочей среды, функционально может быть
сгруппирован на несколько частей:
1) команды выравнивания последовательностей;
2) команды обнаружения генетических вариантов;
3) команды для изменения обозначения хромосом;
4) фильтрации генетических вариантов;
5) анализ вызванных вариантов.
1. Выравнивание
последовательностей
Все команды, представленные ниже, запускаются в
среде запущенного контейнера staphb/bwa
внутри каталога /data. Если команды
не находят файлы для выполнения, требуется убедиться, что рабочий каталог
верный. Для этого нужно выполнить команду pwd
&& ls –lF, в результате которой должны отображаться файлы вроде PID25001.fastq
и GCF_000001405.40_GRCh38.p14_genomic.fna.
Для индексации референсного генома используется
инструмент bwa index (строка 1),
создающий несколько индексных файлов (например, .amb, .ann, .bwt, .pac, .sa), необходимых
для последующего выравнивания.
Команда bwa mem
выполняет выравнивание последовательности пациента fastq-файла на референсный
геном с добавлением информации о группе прочтений (строка 2).
bwa mem -t 8
-R '@RGID:SMP001LB:LBR001PL:NovaSeq6000PU:NS5001234SM:Human-SampleA'
GCF_000001405.40_GRCh38.p14_genomic.fna
PID25001.fastq > output_PID25001.sam
Объяснение элементов команды:
-t 8 – опция задает число
потоков процессора (8 ядер CPU) для ускоренного параллельного выполнения
алгоритма. Для другого процессора это будет другим числом.
-R
'@RGID:SMP001LB:LBR001PL:NovaSeq6000PU:NS5001234SM:Human-SampleA' – опция -R добавляет блок @RG (сокращение от Read Group – группа прочтений)
в выходной файл sam. Блок @RG – это часть
заголовочной секции в файле формата BAM или SAM, которая содержит важную
информацию о происхождении, характеристиках и обстоятельствах получения группы
прочтений. Если в нем ничего не указано, то опция -R позволяет
вставить пользовательские значения. Даже если в файле есть секция с информацией
о группах прочтений, она будет перезаписана этими значениями через аргумент -R. Ниже перечислены задаваемые значения
блока @RG, похожие на реальные, которые часто используются в исследовательских
проектах и публикациях.
ID:SMP001 – идентификатор
группы прочтений. Обычно состоит из буквенно-цифрового префикса и номера,
уникального для каждого эксперимента.
LB:LBR001 – название
библиотеки. Часто также имеет числовой суффикс, позволяющий различать отдельные
партии библиотек. Библиотека – это ДНК-пробы одного пациента или индивида,
подготовленные специально для последующего секвенирования и анализа.
PL:NovaSeq6000 – современная
платформа секвенирования, выпущенная Illumina в 2017 г. Другие возможные
варианты включают HiSeq X Ten, NextSeq, MiSeq и т.п.
PU:NS5001234 – уникальное
имя платформы или прибора. Может включать серийный номер машины, код площадки
или любое другое уникальное сочетание символов.
SM:Human-SampleA – имя образца (вид организма и наименование пробы) может
содержать идентификатор (PID), номер конкретного пациента или источник
биоматериала. В условиях необходимости персонификации данных пример может выглядеть
так: HS-SampleA → Пациент Петров В.В. или HS-SampleB → Пациент Иванов В.В.
(прим., здесь HS от HomoSapiens).
GCF_000001405.40_GRCh38.p14_genomic.fna – путь
к файлу референсного генома, на который будут производиться выравнивания.
Обычно это последовательность в формате FASTA, представляющая человеческий
геном.
PID25001.fastq – путь
к файлу с прочтениями (fastq-файл), которые нужно выровнять на указанный
референсный геном.
output_PID25001.sam – файл
текстового формата SAM (Sequence Alignment Map) для хранения результатов
выравнивания последовательностей. Он включает информацию о том, как прочитанные
фрагменты ДНК были сопоставлены с референсным геномом, положения на референсе,
качество выравнивания и прочие характеристики.
Обратите внимание, что fastq-файл образца здесь
имеет имя PID25001.fastq. У
пользователя будет использоваться другой файл, соответственно, с другим именем.
И далее по коду в названиях файлов имя образца PID25001 должно быть заменено на свое.
Так как директория bind на хост-компьютере связана с директориями /data контейнеров, то, добавив файл output_PID25001.sam в любую из них, он
будет доступен всем контейнерам в своих каталогах /data.
2. Обнаружение
генетических вариантов
Все команды, представленные ниже, запускаются в
среде запущенного контейнера gatk_bcftools
внутри каталога /data. Файл output_PID25001.sam
имеет
большой размер из-за текстового формата и обычно используется как
промежуточный файл. Далее его сжимают в бинарный формат BAM с помощью инструмента
samtools (строка 3), который по умолчанию входит в состав Docker-образа gatk, и, соответственно, в созданный
нами контейнер gatk_bcftools. Затем
производится сортировка bam-файла по координатам (строка 4) и создание индекса
для сортированного bam-файла (строка 5) и просмотр его заголовочной секции
(строка 6).
Далее производятся подготовительные операции с
референсным геномом: индексация и создания словаря индексного генома. Индекс
(строка 7) необходим для быстрого доступа к участкам референсного генома. Сам
индекс сохраняется в файле с расширением .fai,
который создается рядом с исходным fasta-файлом. Команда в строке 8 формирует
словарь из референсного генома, который необходим для обнаружения (вызова)
вариантов.
Для вызова генетических вариантов (строка 9)
используется инструмент HaplotypeCaller из пакета GATK (Genome Analysis
Toolkit).
gatk HaplotypeCaller
-R GCF_000001405.40_GRCh38.p14_genomic.fna
-I sorted_PID25001.bam
-O variants_PID25001.vcf
--native-pair-hmm-threads 8
Команда gatk
запускает HaplotypeCaller для поиска
генетических вариантов в NGS-данных
и сохранения результатов в файл формата VCF.
gatk запускает Genome Analysis Toolkit; HaplotypeCaller – это
конкретный инструмент в GATK, предназначенный для обнаружения генетических
вариантов (однонуклеотидные полиморфизмы – SNPs и короткие вставки / делеции
– Indels).
-R – флаг,
указывающий путь к референсному геному.
GCF_000001405.40_GRCh38.p14_genomic.fna
– это файл референсного генома человека (версия GRCh38, патч p14), который
используется для сопоставления и поиска генетических вариантов.
-I – флаг,
указывающий путь к файлу выравнивания (aligned reads).
sorted_PID25001.bam
– это отсортированный bam-файл, содержащий выровненные прочтения
секвенирования.
-O – флаг,
указывающий выходной файл.
variants_PID25001.vcf
– это файл в формате VCF, куда будут записаны найденные генетические варианты
(SNPs и Indels). Для увеличения скорости выполнения этой команды можно
увеличить количество потоков по числу физических ядер процессора (в конкретном
случае 8) с помощью параметра «native-pair-hmm-threads».
Результат выполнения этой команды – файл с «сырыми»
вызванными вариантами variants_PID25001.vcf.
3. Изменение обозначения хромосом
При создании vcf-файла используются следующие
идентификаторы последовательностей: NC обозначает
официальные референсные хромосомы; NW
– последовательности, полученные методом whole-genome shotgun; NT – неопубликованные или
неклассифицированные последовательности. Для последующей обработки необходимо,
чтобы названия хромосом человека имели вид chr1,…, chrX, chrY, chrMT. Для переименования в стандартные обозначения хромосом
(например, из NC_000023.11 в chrX) используется пакет bcftools (табл. 4).
Для получения уникальных идентификаторов
референсной последовательности из полученного vcf-файла необходимо запустить
команду bcftools query -f с аргументами (строка 10, табл. 3). Выделив в терминале полученный список
идентификаторов хромосом, следует скопировать и вставить его в простой
текстовый редактор. Из списка удаляются все идентификаторы кроме официальных
референсных хромосом, начинающихся на NC.
Для автоматизации процесса переименования хромосом создается
файл rename_NCToChr.txt в папке bind
(соответственно /data внутри
запущенного контейнера) и вручную (при желании можно использовать шаблон для
замены) вводятся строки с соответствиями: старое название <пробел> новое
название:
NC_000001.11
chr1
…
NC_000023.11
chrX
NC_000024.10
chrY
NC_012920.1
chrMT
Используя функцию bcftools annotate --rename-chrs (строка 11), все хромосомы переименуются в соответствии с вышеназванной схемой и будет создан файл renamed_variants_PID25001.vcf.
Таблица 4. Список
хромосом и их идентификаторов в базе данных NCBI для референсного генома в файле GCF_000001405.40_GRCh38.p14_genomic.fna
Хромосома | RefSeq |
1 | NC_000001.11 |
2 | NC_000002.12 |
3 | NC_000003.12 |
… | … |
22 | NC_000022.11 |
X | NC_000023.11 |
Y | NC_000024.10 |
MT | NC_012920.1 |
4.
Фильтрация генетических
вариантов по их качеству, глубине прочтения и др.
заданным параметрам
Чтобы отбросить низкокачественные варианты из
полученного vcf-файла, следует использовать определенные критерии фильтрации
(строка 12). Команда ниже производит фильтрацию вариантов из vcf-файла renamed_variants_PID25001.vcf на основе
нескольких критериев качества и сохраняет результат в файл filt_PID25001_FORMAT-DP.vcf.
bcftools filter -s LowQual -S . -e 'QUAL <
30 || FORMAT/DP < 20 || MQ < 40 || QD < 2'
renamed_variants_PID25001.vcf > filt_PID25001_FORMAT-DP.vcf
-s
LowQual – параметр присваивает метку LowQual отбракованным вариантам.
-S . – оставляет
пустым поле FILTER неотбракованных вариантов.
-e 'QUAL <
30 || FORMAT/DP < 20 || MQ < 40 || QD < 2' – условие, которое помечает
(отбраковывает) варианты с качеством (QUAL) ниже 30; с глубиной прочтения (DP,
depth of coverage) ниже 20; со средним качеством выравнивания (MQ, mapping
quality) менее 40, с отношением качества к глубине (QD, Quality per Depth
ratio) менее 2. Если характеристики варианта соответствуют хотя бы одному из
этих правил, то варианту присваивается метка LowQual, если выше – PASS.
Для того чтобы удалить варианты с пометкой LowQual и оставить только с меткой PASS, используется команда bcftools view с опцией -f
(строка 13).
Таким образом остаются только те варианты, которые
соответствуют критериям качества, указанным в условии фильтра. На выходе этого
этапа получается vcf-файл с отфильтрованными по качеству вариантами — fin_filt_FDP_PID25001.vcf.
5. Анализ вызванных
вариантов в среде R/bioconductor
После того как будет получен vcf-файл, его можно
загрузить в среду программирования R, провести статистический анализ и
аннотацию вариантов. Общий алгоритм анализа вызванных вариантов приведен на рис. 4. Специально для этого исследования
создан R-скрипт VCFtoVarGnomAD.R, который можно скачать по ссылке [6] в виде QR-кода
на рис. 1.
После подготовки вычислительной среды необходимо
осуществить:
1)
извлечение и подготовку данных из vcf-файлов;
2) поиск
информации об обнаруженных вариантах в специализированных базах данных (пример –
gnomAD) и объединение полученной информации;
3)
оптимизацию и улучшение структуры объединенных данных;
4)
финальную фильтрацию по заданным критериям значимости и оценку генетических
вариантов.
Рисунок 4.
Блок-схема конвейера биоинформатических инструментов: аннотация и анализ
вариантов в среде R/Bioconductor
Далее представлено краткое описание комплексного
биоинформатического анализа генетических данных в R-среде из vcf-файла fin_filt_FDP_PID25001.vcf , полученного
ранее.
5.1. Извлечение и подготовка данных из vcf-файлов
Задача заключается в том, чтобы в созданном на
предыдущих этапах vcf-файле выбрать те варианты, которые соответствуют геномным
диапазонам интересующих генов и их транскриптов. Как правило, для удобства
профессиональной коммуникации гены обозначаются сокращенными обозначениями в
виде символов генов (например, «TTN», «BRCA1» и т.д.). Символьные обозначения
генов требуют их конвертации в числовые идентификаторы, более приспособленные
для автоматизированного поиска в специализированных базах данных (например, в
NCBI). Для этого сначала производится подготовка R-среды и инструментов:
устанавливаются и подключаются необходимые пакеты, такие как VariantAnnotation,
AnnotationDbi, Biostrings
и пр., используемые для загрузки vcf-файлов в R и анализа
созданных на основе эти файлов объектов (далее, R-объектов).
Рисунок 4. Блок-схема конвейера биоинформатических инструментов: аннотация и анализ вариантов в среде R/Bioconductor
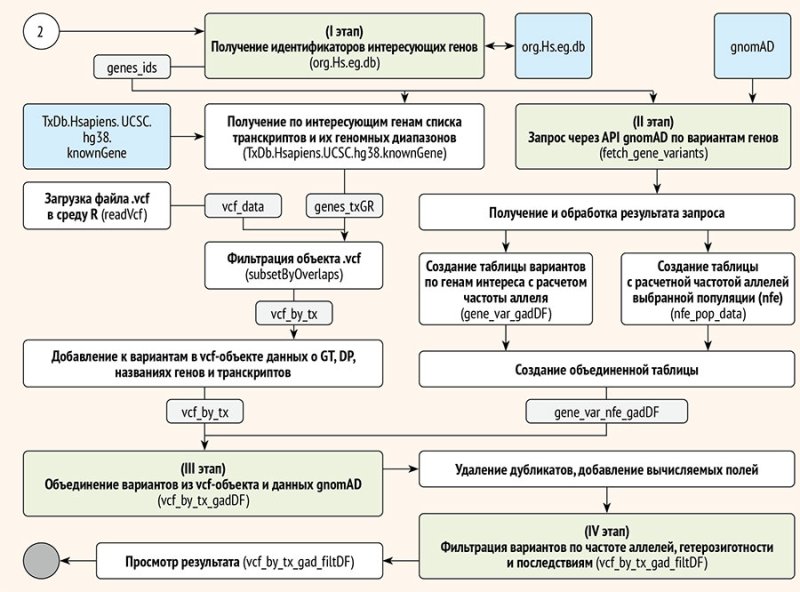
Для конвертации символов генов в уникальные
идентификаторы Entrez ID используется база данных org.Hs.eg.db.
Результатом этой конвертации является перечень идентификаторов genes_ids. По
базе данных транскриптов известных генов TxDb.Hsapiens.UCSC.hg38.knownGene получаются
координаты транскриптов интересующих генов genes_ids
в виде R-объекта геномных диапазонов класса Granges с именем genes_txGR, который можно
использовать далее для отбора вариантов, заданных в исследовании генов и
транскриптов.
Далее в R-среду с использованием функции readVcf() загружается
vcf-файл образца (пациента), полученный на предыдущих этапах, и создается
объект vcf_data. В нем с помощью
функции subsetByOverlaps() отбираются
варианты, соответствующие диапазонам заданных транскриптов genes_txGR.
В vcf-объекте vcf_by_tx добавляются
столбцы с характеристиками каждого варианта, такими как генотип, глубина
покрытия, местоположение и характер мутации. В результате этого получается
полноценная таблица vcf_by_txDF,
отражающая все аспекты каждого конкретного генетического варианта, удобная для
последующих анализов.
5.2.
Поиск информации об обнаруженных вариантах в специализированных базах данных
(например, gnomAD) и объединение полученной информации
Как правило, после обнаружения варианта, следующим
этапом проводится проверка имеющихся сведений о его статистической ассоциации с
болезнями человека. Учитывая статистический характер информации об этой
ассоциации, рекомендуется обращаться в те базы данных, которые накопили
достаточное число случаев, в которых этот вариант присутствует при определенных
болезнях или синдромах, например, gnomAD [7]. Специальные
программные API-интерфейсы (Application Programming Interface) к этим базам
данных позволяют автоматически получать информацию о генетических вариантах для
последующих воспроизводимых аналитических исследований. Ниже приводится
описание важных с клинической и биологической точки зрения характеристик
вариантов, которые можно получить из gnomAD:
аллельная частота, гетерозиготность и последствия варианта.
Распространенность
вариантов или аллельная частота. Редко встречающиеся варианты чаще ассоциируются с
патологией, тогда как широко распространенные варианты обычно считаются
безвредными. На относительную распространенность конкретной аллели (или
варианта) в определенной популяции указывает показатель «частота варианта» или
«аллельная частота». Зная число экземпляров определенной аллели (варианта) в
выборке – ac (от англ. allele count), и полное количество всех
возможных аллелей в рассматриваемом участке генома для всей выборки – an (от англ. allelic number), легко вычислить долю конкретной
аллели в популяции, т.е. аллельную частоту af:
af = ac/an. Необходимо
помнить, что каждая особь имеет две
копии каждого участка хромосомы, поэтому общее количество аллелей
рассчитывается умножением числа образцов на 2.
Значение показателя частоты аллели (gnomAd_AF) считается редким, если оно ниже определенного
порога, обычно равного примерно 0,01%
(1 × 10 − 4) или менее. То есть частота
встречается меньше чем в одном индивиде на каждые 10 тыс человек. Конкретный
порог зависит от контекста и целей исследования, но принято выделять 3 основных
диапазона:
- редкий
вариант: AF < 0,01%; - нечасто
встречающийся вариант: 0,01% ≤ AF < 1%; - распространенный
вариант: AF ≥ 1%.
Эти градации полезны для оценки патогенетической
значимости генетического варианта согласно международным стандартам ACMG / AMP / CLINGEN.
Редко встречающиеся варианты с низкой частотой могут быть кандидатами на роль
патогенных мутаций, особенно если имеются дополнительные подтверждающие
доказательства.
Гетерозиготность. Многие генетические
заболевания проявляются именно в гетерозиготном состоянии (когда пациент несет
одну копию мутантного гена наряду с нормальной копией). Такие состояния важны
для выявления аутосомно-доминантных болезней, гетерозиготных комбинаций в
сцеплении с полом и пр. Определение статуса гетерозиготности позволяет точнее
классифицировать варианты и оценивать их вклад в развитие патологий.
Последствия мутации (варианта). В
API-интерфейсе запроса к gnomAD тип
мутации обозначается полем с оценкой последствия варианта – consequence. Это поле содержит аннотацию
воздействия мутации на структуру и функцию белка или мРНК, полученную с помощью
программы VEP (Variant Effect Predictor). Значения поля consequence могут включать такие значения, как missense_variant – мутация приводит к замене аминокислоты (замена
одной аминокислоты на другую); synonymous_variant
– замена нуклеотида, но сохранение аминокислоты неизменной; frameshift_variant – нарушение рамки
считывания (делеция или вставка, меняющая длину цепочки); stop_gained – появление мутации преждевременного стоп-кодона; start_lost – потеря старт-кодона
трансляции; intron_variant – расположение
варианта в области интрона; splice_region_variant
– нахождение варианта вблизи сайта сплайсинга; intergenic_variant – вариант находится между генами. Критерии на
основе аллельной частоты, гетерозиготности и последствий варианта используются
в R-скрипте на последнем этапе для отбора потенциально значимых с клинической
точки зрения вариантов.
По данным из таблицы vcf_by_txDF производится поиск клинически значимой информации о
вызванных вариантах в базе данных gnomAD.
Для этого производится запрос данных через API-интерфейс gnomAD с помощью специально написанной функции fetch_gene_variants. Для точного и эффективного извлечения
необходимой информации используется запрос в формате GraphQL. Полученные данные обрабатываются, производится поиск и
выделение редких и значимых вариантов, связанных с определенными эффектами
(например, заменами аминокислот, появлением стоп-кодонов). Кроме этого,
производится расчет популяционно-зависимых величин, таких как частота аллелей.
Итоговая таблица gene_var_nfe_gadDF
содержит расширенный перечень сведений о каждом варианте, включающий
локализацию, частотность и функциональное значение варианта.
5.3.
Оптимизация и улучшение структуры объединенных данных
Таблица gene_var_nfe_gadDF
предыдущего этапа содержит лишнюю информацию ввиду объединения разнородных
таблиц. Поэтому необходимо ее очистить и привести к удобочитаемому виду,
устранить избыточные данные и подготовить ее для углубленного анализа. Для
этого обозначения вариантов в gene_var_nfe_gadDF
переводятся в общепринятый формат для gnomAD,
обеспечивающий однозначное соответствие между источниками данных. Производится
удаление повторяющихся данных, устраняются дублирующие или избыточные столбцы.
Для оптимизации структуры данных осуществляется комбинирование ключевых полей и
изменение формы представления, чтобы уменьшить размер итогового набора данных и
повысить удобство его восприятия. В итоге формируется компактная и
информативная таблица vcf_by_tx_gadDF,
идеально подходящая для последующего анализа и клинической интерпретации.
5.4.
Финальная фильтрация и оценка генетических вариантов
Задача этого этапа заключается в том, чтобы выбрать
самые значимые генетические варианты, основываясь на жестких критериях, и
подготовить отчет, пригодный для медицинского анализа и диагностики. В примере,
приведенном в R-скрипте, в итоговую таблицу gene_var_nfe_gadDF
отбирались варианты по определенным критериям:
1) наличие гетерозиготности (значение
«гетерозиготный»);
2) аллельная частота <1%, а для близкой нам
популяции, например, население северной Европы (non-finnish europeans, NFE) – значение af_nfe < 0,01;
3)
со значимыми последствиями мутаций (поле consequence имело значение missense_variant, stop_gained, frameshift_variant, start_lost).
В ходе фильтрации вариантов производятся следующие
действия:
1) фильтрация записей с вариантами по заданным
параметрам с использованием функций пакета dplyr;
2) проверка и вывод результатов функции head(),
чтобы удостовериться в правильности определения выводимых полей (столбцов)
данных.
В результате описанных этапов создается таблица vcf_by_tx_gad_filtDF, содержащая
значимые варианты, идентифицированные и готовые для дальнейшего глубокого
анализа и медицинских выводов (табл. 5).
Таблица 5. Результаты
медико-генетического исследования данных экзома пациента
Вариант (по (hg38)) | Зиготн. | Ген | Транскрипт | cDNA изменение | АК замена | DP |
chr2:44312653T>C | гет. | SLC3A1 | ENST00000260649 | c.1400T>C | p.Met467Thr | 176 |
chr2:178795157GT>G | гет. | TTN | ENST00000342175 | c.1009delA | p.Thr337fs | 242 |
chr4:44691731CT>C | гет. | GUF1 | ENST00000281543 | c.1548delT | p.Pro517fs | 83 |
chr8:73981555CAAAG>C | гет. | TMEM70 | ENST00000312184 | c.720_723delAGAA | p.Glu241fs | 114 |
chr16:88714481CCG>C | гет. | CTU2 | ENST00000453996 | c.1198_1199delGC | p.Ala400fs | 141 |
chr20:10405526C>T | гет. | MKKS | ENST00000347364 | c.1434G>A | p.Trp478* | 286 |
Примечание. Зиготн. – зиготность, гет. – гетерозигота,
АК замена – аминокислотная замена, DP – глубина прочтений.
Все представленные мутации являются
гетерозиготными, т. е. присутствуют одна нормальная аллель и одна мутированная.
Большинство мутаций приводят к сдвигу рамки считывания (frameshift mutations) –
это наиболее серьезные изменения, способные существенно повлиять на структуру
белка. Одна мутация (MKKS) приводит к преждевременному появлению стоп-кодона,
что также нарушает синтез функционального белка. Значительная глубина прочтений
подтверждает надежность результатов секвенирования. Эти данные могут
использоваться для дальнейших исследований генетики пациента, диагностики
наследственных заболеваний или оценки риска развития патологий.
Для валидации всего представленного алгоритма
обнаружения и аннотации вариантов использовались результаты
медико-генетического анализа, полученные в сертифицированной
медико-генетической лаборатории. Полученные результаты оказались идентичными.
Выводы
Использование предложенного алгоритма и программного
кода позволяет получить на персональных компьютерах без проприетарных программ
варианты геномной последовательности (экзома) пациента, пригодные для
последующего анализа и интерпретации. Разработка кода выполнена совместно с
использованием системы искусственного интеллекта GigaChat (версия 07.06.2025) и
проверена в средах контейнеров bwa, gatk c bcftools, bioconductor, RStudio под
ОС Ubuntu 24.04.2 LTS на процессоре AMD Ryzen™ 7 5800U with Radeon™ Graphics ×
16 с 32 Гб ОЗУ, 1,0 Tb SSD. Несмотря на доступность, по нашему опыту,
необходимо учитывать некоторые нюансы его практического использования.
- Требования к оборудованию. Анализ данных NGS
возможен на обычных офисных или домашних компьютерах, однако их
производительность является важным фактором. Еще одно значительное ограничение
в использовании этих инструментов – наличие Linux-операционных систем, которые
врачами часто негативно воспринимаются и требуют дополнительного освоения. - Время обработки данных. Несмотря на доступность оборудования, время обработки NGS-данных
остается значительным. Например, обработка экзома одного образца требует
примерно 2–3 ч вычислительного времени, что позволяет обрабатывать данные
максимум 3–4 пациентов на одном компьютере в течение рабочего дня. Предварительная
настройка инструментов и библиотек может занимать около 1–2 ч при высокой
скорости подключения к Интернету. - Исследовательская
природа анализа. Сегодня процесс выявления значимых генетических вариантов
на основе NGS-данных относится скорее к сфере исследований, а не рутинной процедурой медицинского
обслуживания. Однако распространение и упрощение этих технологий могут
позволить врачам самостоятельно интерпретировать полученные генетические данные
пациентов, подобно тому, как они стали уверенно читать результаты компьютерной
томографии (КТ), магнитно-резонансной томографии (МРТ), электрокардиограммы
(ЭКГ) и других диагностических методов. - Образовательная
ценность метода. Использование NGS-данных пациента для самостоятельного
анализа имеет большой образовательный потенциал. Осваивая методики анализа, медицинские
специалисты приобретают новые знания и умения, формируют сообщество
профессионалов, способствующих распространению инноваций и увеличению
количества квалифицированных специалистов в данной области. - Роль
врачей в популяризации биоинформатики. Активное распространение и
обучение основам биоинформатического анализа среди медицинских работников ведет
к улучшению самого процесса анализа NGS-данных и повышает качество диагностики
заболеваний, связанных с генетическими изменениями. - Использование
систем искусственного интеллекта. Развитие искусственных помощников
коренным образом расширяет возможности биоинформатики и медицины. Тем не менее врачи
остаются ключевыми фигурами в постановке вопросов, формировании рабочих гипотез
и принятии важных клинических решений в контексте реальных потребностей
пациента.
Важным остается вопрос определения необходимого
объема специализированных знаний по биоинформатике и оптимального этапа
профессионального образования, на котором врач должен осваивать соответствующие
компетенции. Уже сейчас очевидно, что в дилемме «нужна / не нужна»
биоинформатика врачу – ответ однозначен. Еще в 2001 г. на страницах журнала Science среди
ведущих экспертов отрасли звучало мнение о неизбежности интеграции
биоинформатики в медицинскую практику. При опросе о перспективах научной
карьеры и о том, кто будет заниматься биоинформатикой, Грэм Кэмерон,
соруководитель Европейского института биоинформатики, предрек: «Вы все
будете, так что вам лучше поторопиться». Сегодня этот прогноз сбывается в
полной мере
[35]. [ПМ1]
[1]
Репозиторий с исходными данными и скриптами проекта BioVarExplorer. Режим
доступа: https://github.com/medRwork/BioVarExplorer.
[2]
Референсный геном человека из базы данных Refseq National Center for Biotechnology Information
– NCBI. Режим доступа: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.40/.
[3]
Портал GitHub. Режим доступа: https://github.com/medRwork/BioVarExplorer.
[4]
Docker
Docs. Режим
доступа: https://docs.docker.com/
[5] Bioconductor – Docker for
Bioconductor. Available at: https://bioconductor.org/help/docker/.
[6]
Репозиторий с исходными данными и скриптами проекта BioVarExplorer. Режим
доступа: https://github.com/medRwork/BioVarExplorer.
[7]
База данных агрегации генома gnomAD. Режим доступа: https://gnomad.broadinstitute.org/.
Список литературы / References
Развернуть
- Vicente AM, Ballensiefen W, Jönsson JI. How personalised medicine will transform healthcare by 2030: the ICPerMed vision. J Transl Med. 2020;18(1):180. https://doi.org/10.1186/s12967-020-02316-w.
- Khan A, Barapatre AR, Babar N, Doshi J, Ghaly M, Patel KG et al. Genomic medicine and personalized treatment: a narrative review. Ann Med Surg. 2025;87(3):1406-1414. https://doi.org/10.1097/MS9.0000000000002965.
- Desmond-Hellmann S. Toward precision medicine: a new social contract? Sci Transl Med. 2012;4(129):129ed3. https://doi.org/10.1126/scitranslmed.3003473.
- Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793-795. https://doi.org/10.1056/NEJMp1500523.
- Manolio TA, Narula J, Bult CJ, Chisholm RL, Deverka PA, Ginsburg GS et al. Genomic medicine year in review: 2023. Am J Hum Genet. 2023;110(12):1992-1995. https://doi.org/10.1016/j.ajhg.2023.11.001.
- Beg A, Parveen R. Role of bioinformatics in cancer research and drug development. In: Translational Bioinformatics in Healthcare and Medicine. Elsevier. 2021;141-148. https://doi.org/10.1016/b978-0-323-89824-9.00011-2.
- Attwood TK, Blackford S, Brazas MD, Davies A, Schneider MV. A global perspective on evolving bioinformatics and data science training needs. Brief Bioinform. 2019;20(2):398-404. https://doi.org/10.1093/bib/bbx100.
- Chatterjee A, Ahn A, Rodger EJ, Stockwell PA, Eccles MR. A guide for designing and analyzing RNA-Seq data. In: Raghavachari N, Garcia-Reyero N, eds. Gene Expression Analysis. Methods MolBio. 2018;1783:35-80. https://doi.org/10.1007/978-1-4939-7834-2_3.
- Kolanu ND. CRISPR-Cas9 Gene Editing: Curing Genetic Diseases by Inherited Epigenetic Modifications. Glob Med Genet. 2024;11(1):113-122. https://doi.org/10.1055/s-0044-1785234.
- Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: An overview. Hum Immunol. 2021;82(11):801-811. https://doi.org/10.1016/j.humimm.2021.02.012.
- Toriello HV, Smith SD. Hereditary Hearing Loss and Its Syndromes. 3rd ed. Oxford University Press; 2013. Available at: https://global.oup.com/academic/product/hereditary-hearing-loss-and-its-syndromes9780199731961?cc=ru&lang=en&.
- Owusu Obeng A, Fei K, Levy KD, Elsey AR, Pollin TI, Ramirez AH et al. Physician-Reported Benefits and Barriers to Clinical Implementation of Genomic Medicine: A Multi-Site IGNITE-Network Survey. J Pers Med. 2018;8(3):24. https://doi.org/10.3390/jpm8030024.
- Carroll JC, Makuwaza T, Manca DP, Sopcak N, Permaul JA, O’Brien MA et al. Primary care providers’ experiences with and perceptions of personalized genomic medicine. Can Fam Physician. 2016;62:e626-e635. Available at: https://www.cfp.ca/content/62/10/e626.
- Campion M, Goldgar C, Hopkin RJ, Prows CA, Dasgupta S. Genomic education for the next generation of health-care providers. Genet Med. 2019;21(11):2422-2430. https://doi.org/10.1038/s41436-019-0548-4.
- Kohzaki H. A proposal for clinical genetics (genetics in medicine) education for medical technologists and other health professionals in Japan. Front Public Health. 2014;2:128. https://doi.org/10.3389/fpubh.2014.00128.
- Kudron EL, Deininger KM, Aquilante CL. Are Graduate Medical Trainees Prepared for the Personalized Genomic Medicine Revolution? Trainee Perspectives at One Institution. J Pers Med. 2023;13(7):1025. https://doi.org/10.3390/jpm13071025.
- Lee-Barber J, Kulo V, Lehmann H, Hamosh A, Bodurtha J. Bioinformatics for medical students: a 5-year experience using OMIM® in medical student education. Genet Med. 2019;21(2):493-497. https://doi.org/10.1038/s41436-018-0076-7.
- Schwartz JE, Ko P, Freed S, Safdar N, Christman M, Page R et al. Integrating an interprofessional educational exercise into required medical student clerkships - a quantitative analysis. BMC Med Educ. 2025;25(1):168. https://doi.org/10.1186/s12909-024-06499-4.
- Hoffman JD, Thompson R, Swenson KB, Dasgupta S. Complexities of Clinical Genetics Consultation: An Interprofessional Clinical Skills Workshop. MedEdPORTAL. 2020;16:10869. https://doi.org/10.15766/mep_2374-8265.10869.
- Kopel J, Brower GL. Perspectives on Consumer and Clinical Genetic Testing Education among Medical Students in West Texas. J Community Hosp Intern Med Perspect. 2022;12(3):28-32. https://doi.org/10.55729/2000-9666.1050.
- Kopel J. Clinical genetic testing in medical education. Proc. 2019;32(1):165-166. https://doi.org/10.1080/08998280.2018.1528937.
- Nightingale KP, Bishop M, Avitabile N, Simpson S, Freidoony L, Buckley S, Tatton-Brown K. Evaluation of the Master’s in Genomic Medicine framework: A national, multiprofessional program to educate health care professionals in NHS England. Genet Med. 2025;27(1):101277. https://doi.org/10.1016/j.gim.2024.101277.
- French EL, Kader L, Young EE, Fontes JD. Physician Perception of the Importance of Medical Genetics and Genomics in Medical Education and Clinical Practice. Med Educ Online. 2023;28(1):2143920. https://doi.org/10.1080/10872981.2022.2143920.
- McCorkell G, Nisselle A, Halton D, Bouffler SE, Patel C, Christodoulou J et al. A national education program for rapid genomics in pediatric acute care: Building workforce confidence, competence, and capability. Genet Med. 2024;26(10):101224. https://doi.org/10.1016/j.gim.2024.101224.
- McGrath SP, Walton N, Williams MS, Kim KK, Bastola K. Are providers prepared for genomic medicine: interpretation of Direct-to-Consumer genetic testing (DTC-GT) results and genetic self-efficacy by medical professionals. BMC Health Serv Res. 2019;19(1):844. https://doi.org/10.1186/s12913-019-4679-8.
- Sepulveda JL. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J Mol Diagn. 2020;22(1):3-20. https://doi.org/10.1016/j.jmoldx.2019.08.006.
- Kadri S, Sboner A, Sigaras A, Roy S. Containers in Bioinformatics: Applications, Practical Considerations, and Best Practices in Molecular Pathology. J Mol Diagn. 2022;24(5):442-454. https://doi.org/10.1016/j.jmoldx.2022.01.006.
- Kang K, Yang Y, Wu Y, Luo R. Integrating Large Language Models in Bioinformatics Education for Medical Students: Opportunities and Challenges. Ann Biomed Eng. 2024;52(9):2311-2315. https://doi.org/10.1007/s10439-024-03554-5.
- Корнеенков АА, Янов ЮК, Рязанцев СВ, Вяземская ЕЭ, Астащенко СВ, Рязанцева ЕС. Метаанализ клинических исследований в оториноларингологии. Вестник оториноларингологии. 2020;85(2):26-30. https://doi.org/10.17116/otorino20208502126.
- Корнеенков АА, Рязанцев СВ, Вяземская ЕЭ, Будковая МА. Меры информативности диагностических медицинских технологий в оториноларингологии: вычисление и интерпретация. Российская оториноларингология. 2020;19(1):46-55. https://doi.org/10.18692/1810-4800-2020-1-46-55.
- Корнеенков АА, Левина ЕА, Вяземская ЕЭ, Левин СВ, Скирпичников ИН. Пространственный кластерный анализ в моделировании доступности медицинской помощи пожилым пациентам с нарушениями слуха. Российская оториноларингология. 2021;20(6):8-19. https://doi.org/10.18692/1810-4800-2021-6-8-19.
- Корнеенков АА, Рязанцев СВ, Левин СВ, Храмов АВ, Вяземская ЕЭ, Скирпичников ИН и др. Пространственно-статистический анализ данных о нарушениях слуха у жителей Челябинской области. Российская оториноларингология. 2021;20(3):39-50. https://doi.org/10.18692/1810-4800-2021-3-39-50.
- Корнеенков АА, Янов ЮК, Вяземская ЕЭ, Медведева АЮ. Вопросы интеграции медицинских биоинформатических технологий в оториноларингологию: проблемы и программные решения. Российская оториноларингология. 2024;23(6):8-19. Режим доступа: https://www.elibrary.ru/kevooo.
- Корнеенков АА, Янов ЮК, Дворянчиков В.В., Вяземская ЕЭ, Медведева АЮ. Медицинская биоинформатика: базовые операции с нуклеотидными последовательностями в программной среде R. Российская оториноларингология. 2025;24(4):13-27. (In Russ.) https://doi.org/10.18692/1810-4800-2025-4-13-27.
- Taylor C. The Future of Bioinformatics. Science. 2000;8. https://doi.org/10.1126/article.63850.
Фото:
Chokniti-Studio/FOTODOM/Shutterstoсk

 1
1 2
2 3
3 4
4


Комментарии (0)